
QIAseq FX DNA Library Kit
For whole-genome or metagenomic NGS library preparation, with or without PCR, for hybrid-capture target enrichment on Illumina instruments.
For whole-genome or metagenomic NGS library preparation, with or without PCR, for hybrid-capture target enrichment on Illumina instruments.
Cat no. / ID. 180477
QIAseq FX DNA library preparation kits are designed for fast, enzymatic preparation of high-complexity whole-genome libraries with low GC bias. Fragment size, input amount (20 pg –1 µg) and batch size are customizable, making the kit highly suited for next-generation sequencing (NGS) projects. The all-enzymatic DNA fragmentation takes just 2.5 hours and requires no DNA shearing equipment, saving substantial time and costs.
Without the use of expensive DNA shearing equipment, substantial time and costs are saved. High-fidelity amplification can be added to the otherwise PCR-free protocol to increase library yield. The streamlined, optimized protocol can be automated, reducing hands-on time and run-to-run variability. The technology is compatible with Illumina sequencing instruments, providing configurations for up to 1536 samples.
The NGS library preparation kit supports researchers working with limited or degraded DNA – including low-input clinical, environmental and microbial samples – where mechanical shearing is impractical or causes sample loss. The kit is equally suited for core labs requiring consistent, automated library preparation across human whole genome sequencing (WGS), metagenomic library preparation and microbial genome sequencing projects.
| Feature | Benefits |
|---|---|
| Enzymatic fragmentation | Compared to mechanical fragmentation:
Sequence-independent fragmentation No sample clean-up before adapter ligation |
| Controlled fragment size | Improves reproducibility |
| Reduced GC bias | Ensures more uniform genome coverage |
| DNA input | Appropriate for low DNA samples |
| Multiplexing | Enables parallel processing of up to 1536 samples |
| Throughput | Configurations for up to 1536 samples |
| Platform compatibility | Illumina and Element |
Need a quote for your research project or would you like to discuss your project with our specialist team? Contact Us.
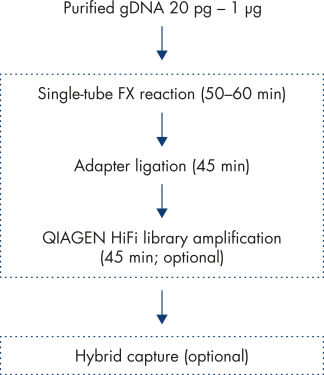
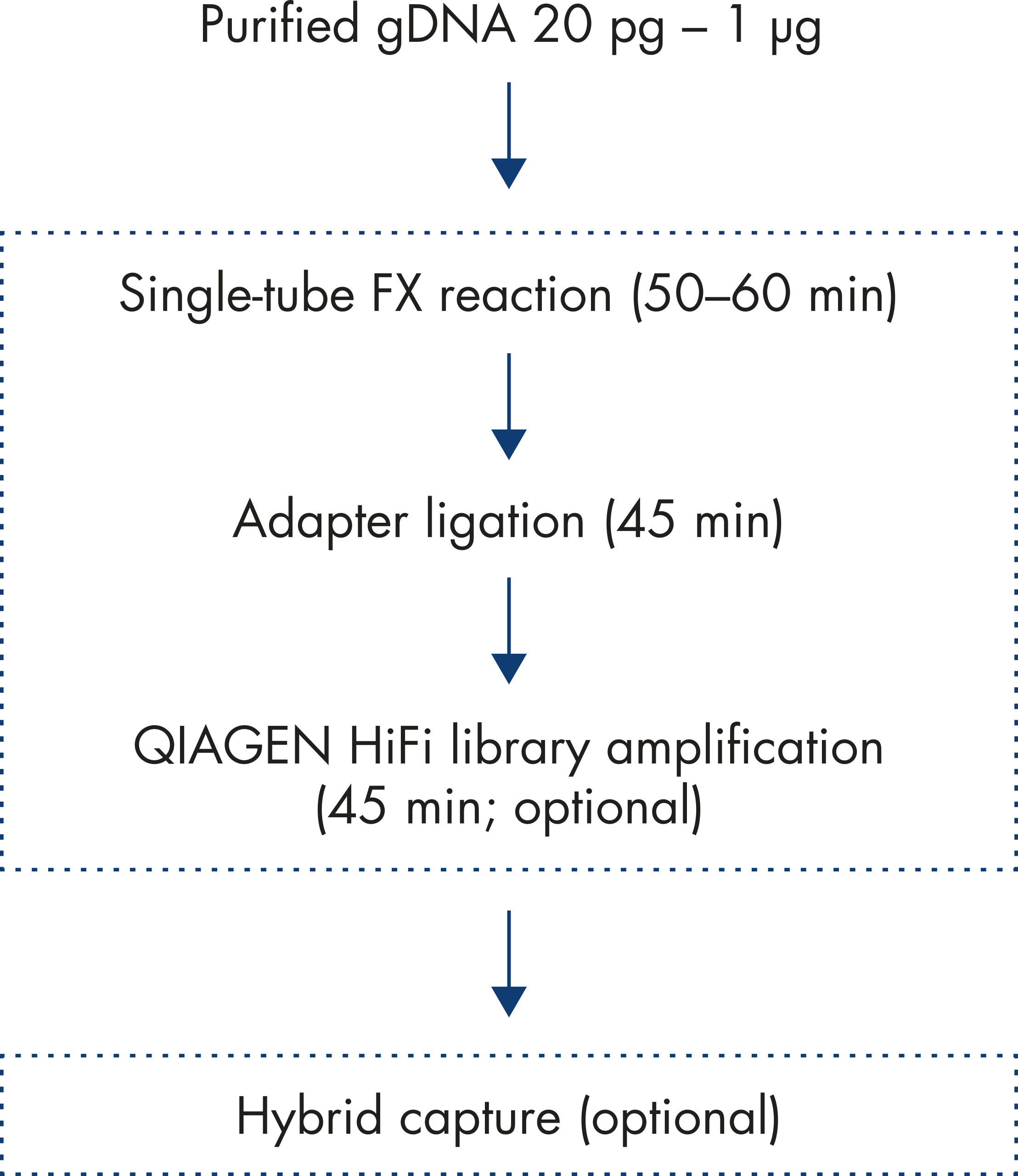
The fast and efficient workflow (see figure: Workflow) for enzymatic library conversion provided by the QIAseq FX DNA Library Kit is specifically designed for success in whole-genome sequencing for a wide range of applications.
| Feature | Specifics | Further details |
|---|---|---|
| Source genomic DNA from any organism | Oil, gut microbiome, wastewater and clinical isolates, etc. | Suitable for metagenomics and profiling whole-genome projects |
| Flexible input range | 20 pg – 1 μg genomic DNA | For low-DNA samples or for libraries that will be used in several downstream applications |
| Enzymatic fragmentation | Comparable to mechanical shearing | Fast 2.5 hour workflow Easily automated |
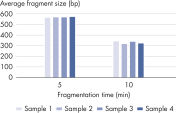
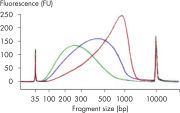
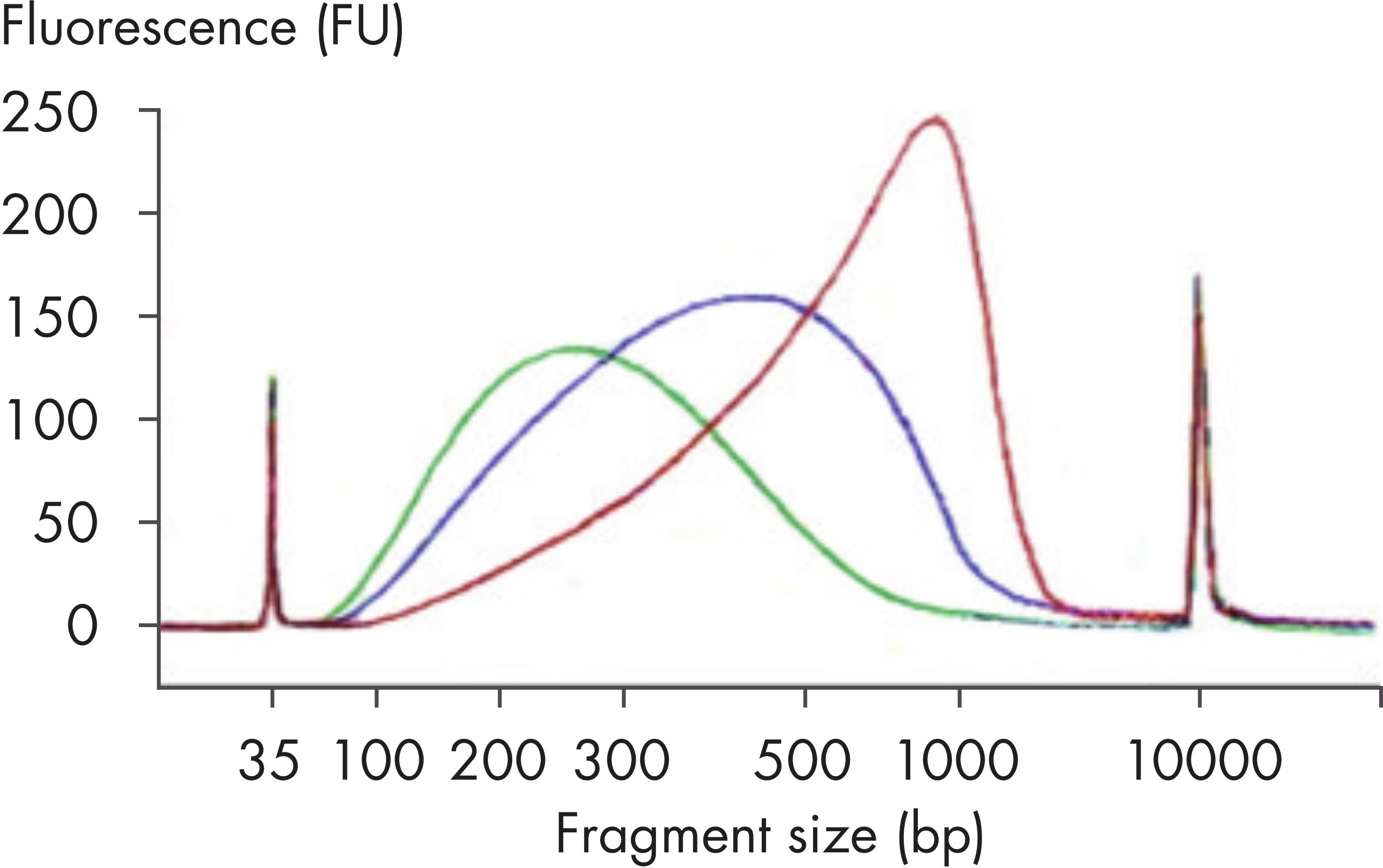
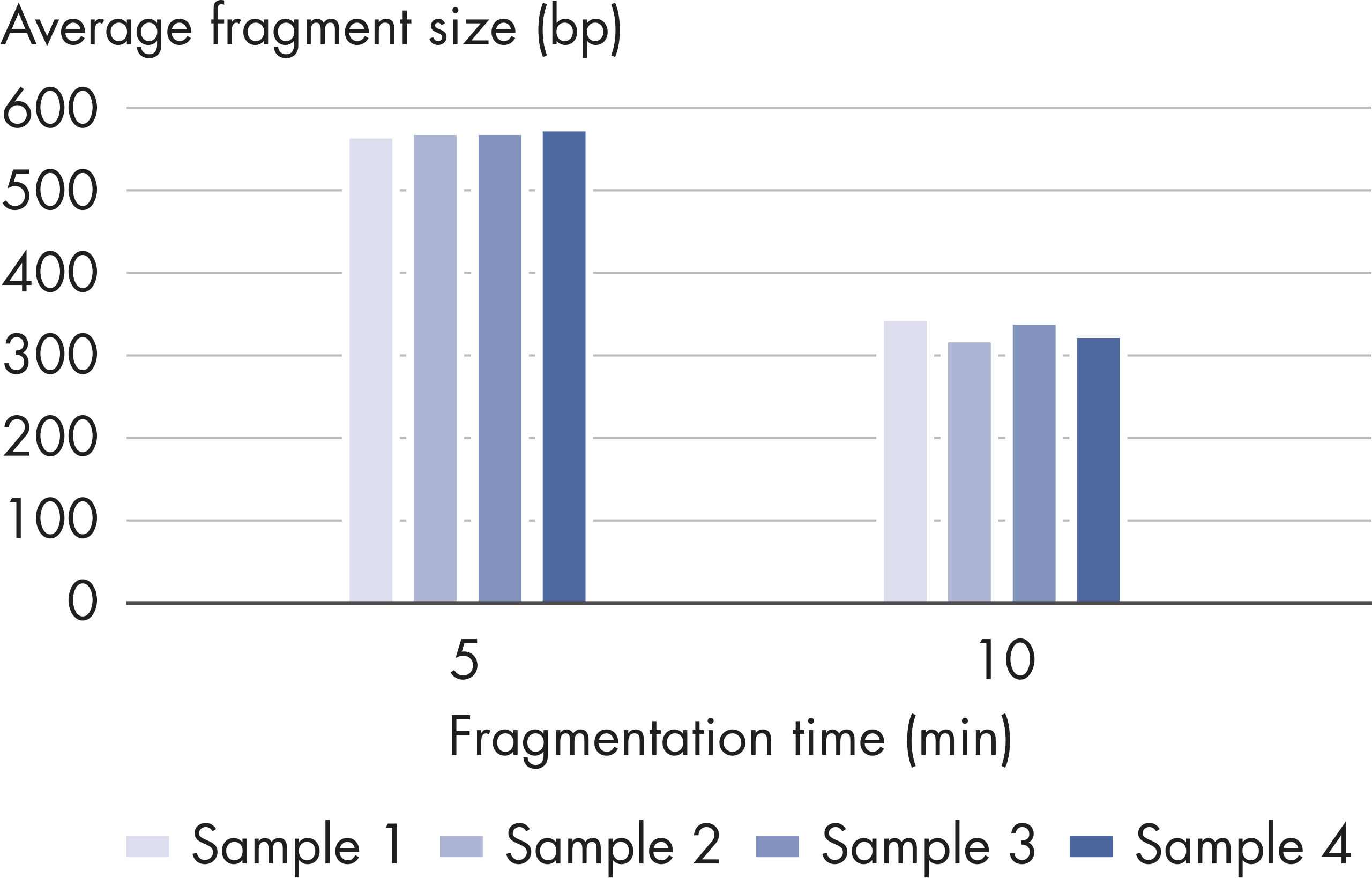
| Customizable fragment size | Highly reproducible fragmentation dependent on incubation time (see figures: Customizable fragment sizes and High reproducibility) |
|
| Similar total coverage depth for genomic targets | No additional sequencing for low-coverage targets | |
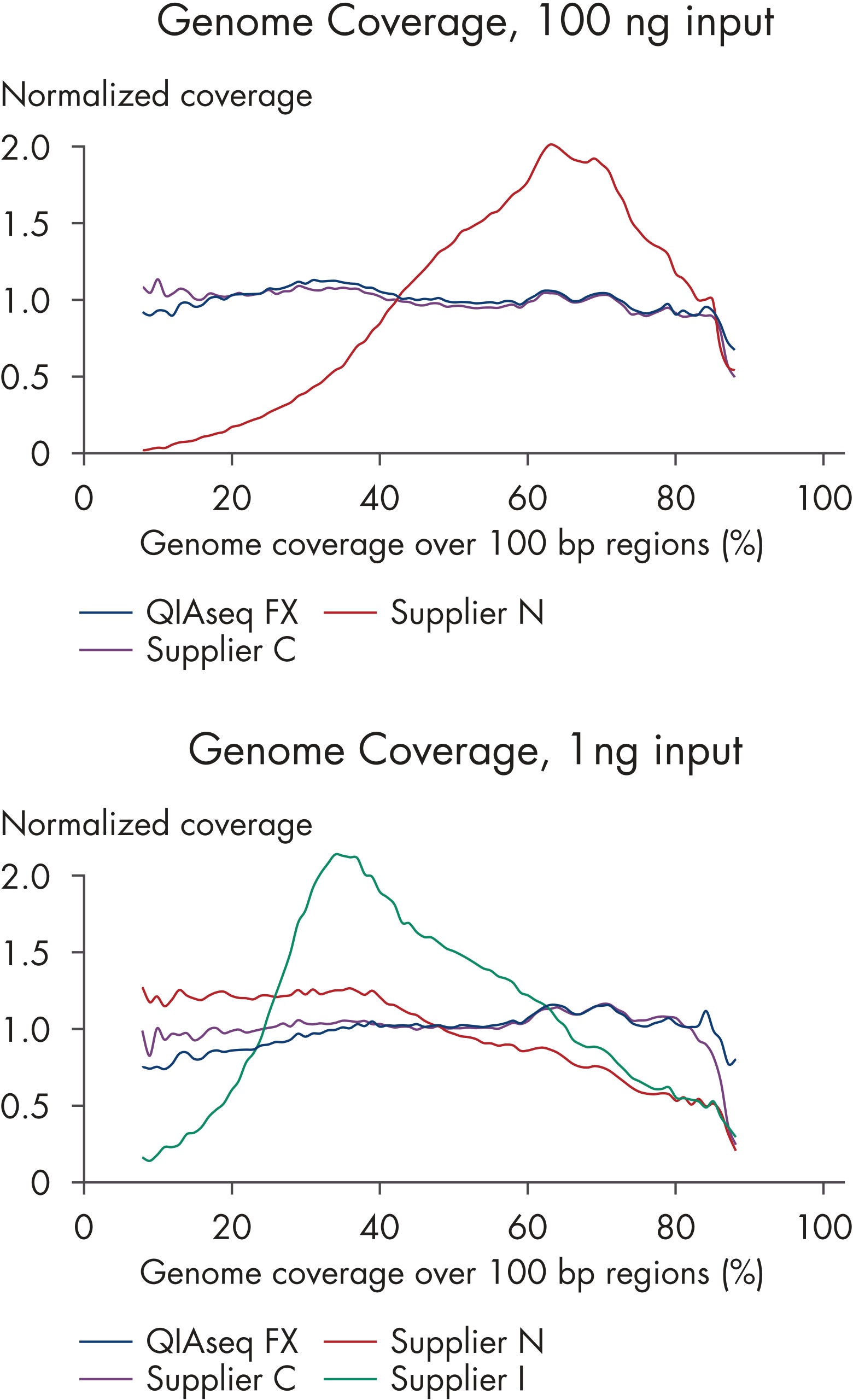
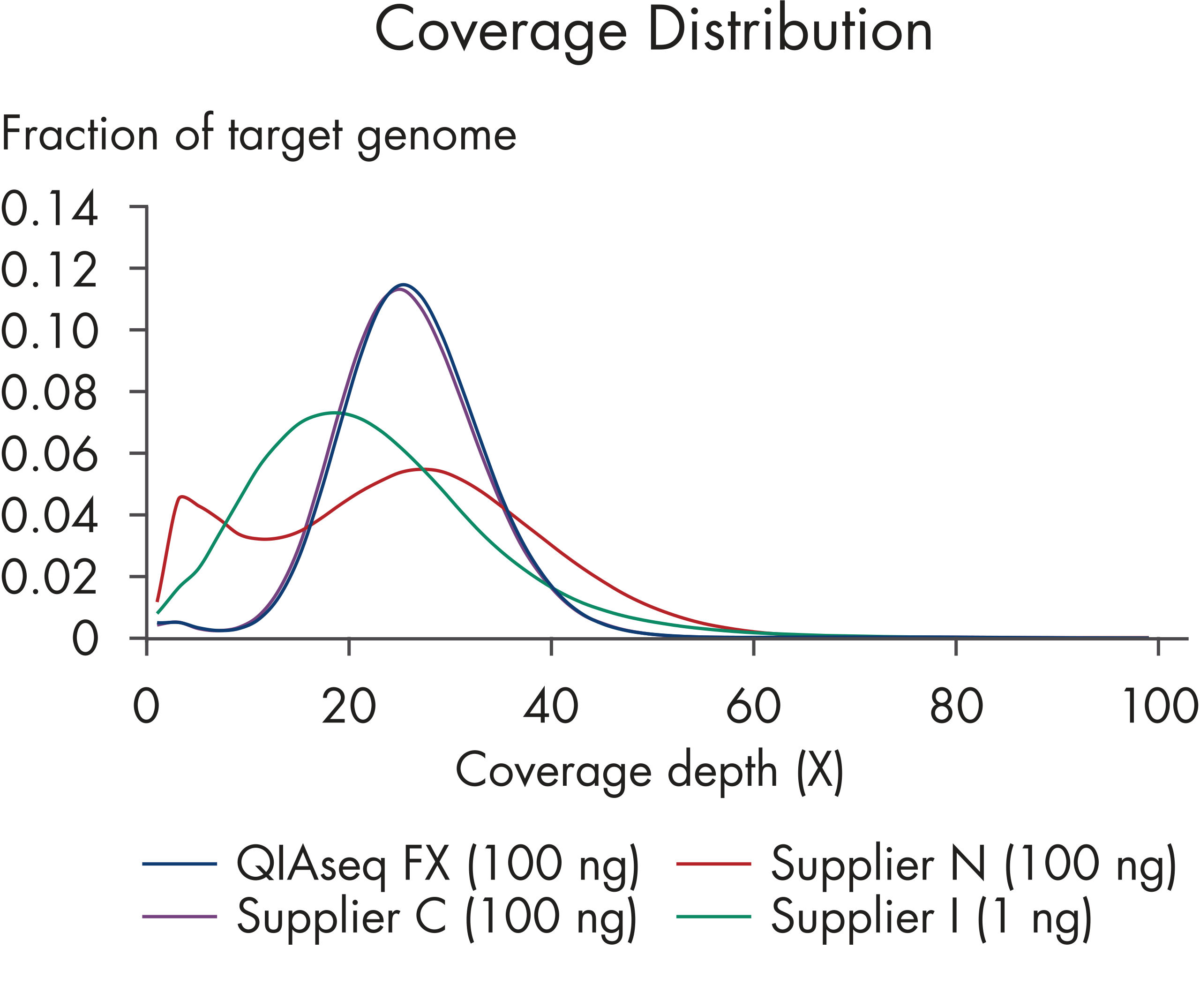
| Minimal GC bias | Improves representation in high- and low-GC microbial genomes (see figures: Minimal GC bias compared to other enzymatic methods) and No significant differences between coverage of high or low GC genomic regions) | |
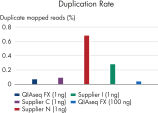
| Coverage | High library complexity with low duplicate rates | Reduces the need for additional sequencing to bring low-coverage targets up to an interpretable coverage range, saving time and resources Maximizes interpretable data and overcomes common shortcoming of other enzymatic fragmentation methods (see figures: Superior coverage distribution and Lower duplication rate) |
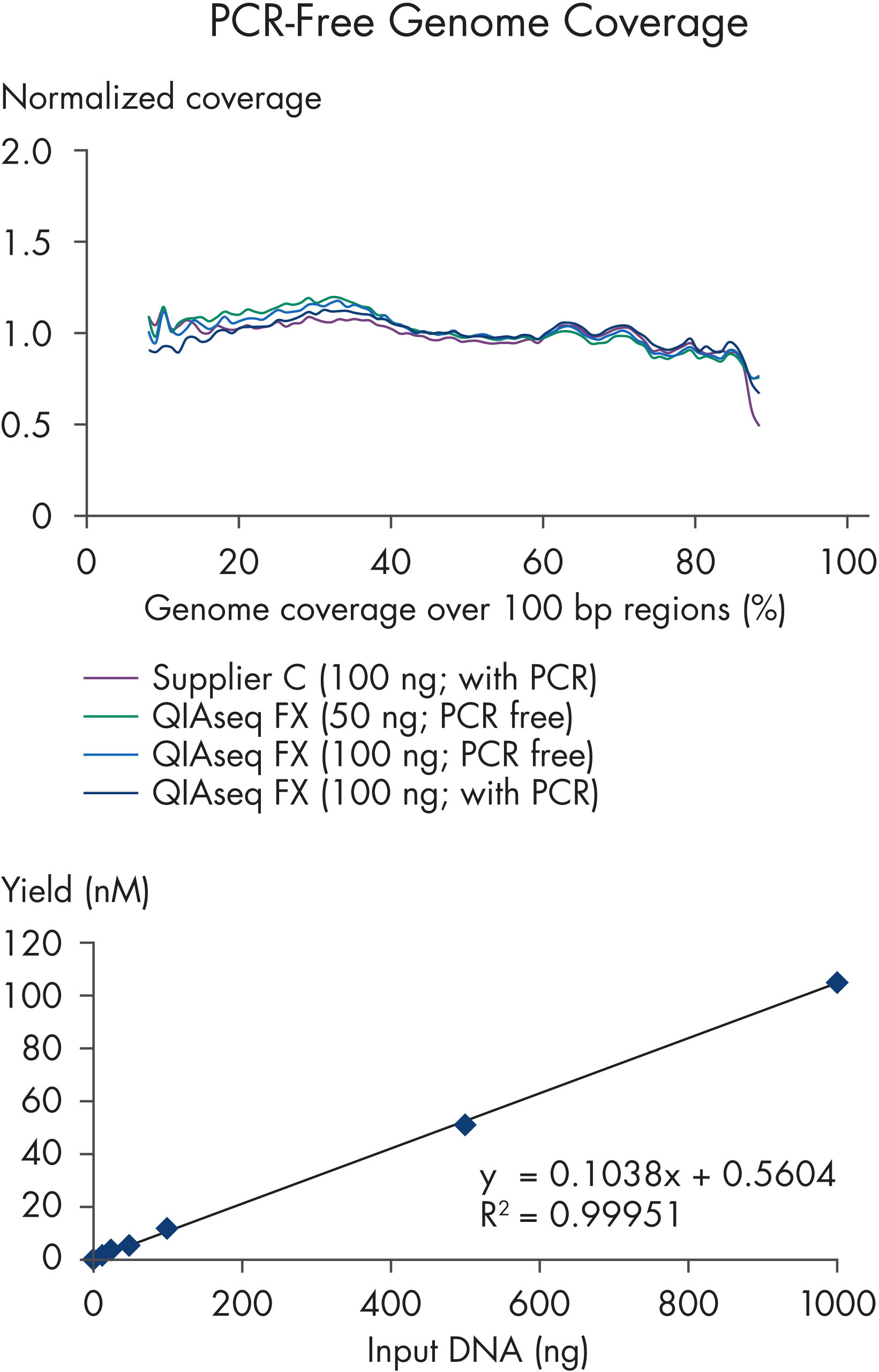
| PCR-free workflow | Adapter ligation chemistry and dual bar coded adapters when PCR amplification is not needed | Provides superior data quality for NGS sequencing of whole-genome libraries |
The novel nuclease formulation, all-enzymatic fragmentation and protocol are optimized for microbial genomes and shotgun (e.g., whole genome, metagenomic) sequencing workflows on Illumina platforms. The fragment size is adjustable simply by changing incubation time of the dsDNA with the nuclease. Optimized enzyme and buffer compositions ensure high sequencing library yield. Streamlined library construction protocols also enable straightforward automation. The fast, fully enzymatic procedure – from DNA fragmentation to NGS library – requires no cleanup steps until after adapters have been ligated to the sample DNA.
Overview
The procedure detailed below consists of three, easy-to-follow steps starting from genomic DNA and ending with sequencer-ready, whole-genome libraries. Dual bar-coded, plate-format adapters are included with the kit. Each well contains a unique combination of two identification bar codes. Up to 1536-plex pooling is possible prior to sequencing.
| Input | Step | Output |
|---|---|---|
| Purified genomic DNA | FX enzymatic fragmentation of genomic or long-range DNA. Fragment size is controlled by the time of the reaction. Sequence-agnostic fragmentation. | DNA fragments of specified size |
| Fragmented DNA | In an end-repair reaction, an A is added to the 3′ ends of the fragmented DNA. | Fragments ready for adapter ligation |
| Platform-specific dual bar coded adaptors are ligated to both end of the DNA fragments; no cleanup necessary. The PCR-free workflow ends here. | Sequencer-ready dual bar-coded whole-genome libraries | |
| Amplification (optional) | In the optional amplification step (only necessary for the PCR workflow), DNA regions with vastly different GC contents are evenly amplified, minimizing sequencing bias caused by PCR. | Increased library yield |
Cleanup and removal of adapter-dimers
Following library construction, the reaction cleanup and removal of adapter dimers can be achieved by using QIAseq Beads, enabling easy automation on various high-throughput automation platforms.
Library normalization
The QIAseq Library Normalizer seamlessly integrates with our protocol, removing the need for tedious qPCR and manual dilution of libraries before pooling. Normalized libraries are ready-to-sequence dsDNA at approximately 4 nmol/L.
QIAseq FX DNA Library Kit generates high-quality whole-genome libraries appropriate for the following types of sequencing:
These types of sequencing are used in the following applications:





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}