

Want to try this solution for the first time?
Get in touch with our team today and request a quote to trial the QIAseq xHYB Long Read Panel (24).
QIAseq xHYB Long Read Panel (24)
Cat no. / ID. 334322
xHYB probe panel for target enrichment for long-read sequencing, fixed panel for 24 samples
Configure at GeneGlobe To see pricing
PanelReagent
QIAseq xHYB Long Read Panel (24)
QIAseq xHYB Long Read Reagent Kit E (24)
QIAseq xHYB Long Read Reagent Kit S (24)
QIAseq xHYB Long Read Panel (96)
QIAseq xHYB Long Read Reagent Kit E (96)
QIAseq xHYB Long Read Reagent Kit S (96)
QIAseq xHYB Long Read Custom Panel (96)
QIAseq xHYB Long Read Panels are intended for molecular biology applications. These products are not intended for the diagnosis, prevention, or treatment of a disease.
Want to try this solution for the first time?
Get in touch with our team today and request a quote to trial the QIAseq xHYB Long Read Panel (24).
Useful links
Below you can find a set of references related to QIAseq Targeted DNA Pro Panels
| Popular Products | GeneGlobe ID |
|---|---|
| QIAseq xHYB Long Read Hereditary Cancer Panel | LXHS-3200Z |
| QIAseq xHYB Long Read HLA Typing Panel | LXHS-200Z |
Features
- High completeness and uniformity of coverage: Achieve comprehensive and even coverage across targeted regions for accurate variant detection
- Gold-standard hybrid capture: Ensures high specificity and sensitivity for target enrichment
- Long read length: Span complex and repetitive genomic regions with sufficient read length for accurate variant calling and phasing
- Flexible and customizable: Tailor panels to specific research needs and budgets
- Optimized for long-read platforms: Specifically designed for use with Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT)
Product Details
QIAseq xHYB Long Read Panels offer highly specific and sensitive hybrid capture-based target enrichment for long-read sequencing, ensuring complete and uniform coverage across complex genomic regions, including high-GC content and structural variations.
These panels are optimized for PacBio (Revio and Vega long-read systems) and ONT long-read systems and provide high-quality data for accurate and reliable downstream analyses. Their flexibility and customization options allow researchers to tailor experiments to their specific needs and budgets, while an optimized, user-friendly workflow ensures ease of use.
Designed to advance cancer research, precision medicine and structural variant analysis, these panels empower scientists with comprehensive, unbiased genomic insights.
Contact us for a specific quote for your research project or to discuss your project with our specialist team.
Performance
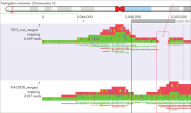
Accurate analysis of high-GC and complex genomic regions
QIAseq xHYB Long Read Panels are designed to overcome the limitations of short-read sequencing by accurately capturing and analyzing regions with high GC content and complex structural variations. These panels use advanced hybrid capture technology to deliver precise and comprehensive genomic insights, making them an essential tool for researchers working with challenging genomic regions.
Gold-standard hybrid capture for high specificity and sensitivity
Unlike amplicon-based approaches, which can introduce bias and inefficiencies, QIAseq xHYB Long Read Panels utilize gold-standard hybrid capture technology to ensure high specificity and sensitivity. This method significantly minimizes off-target reads while providing uniform coverage, even across difficult-to-sequence regions.
Unlocking the power of long-read sequencing for structural variation analysis
Structural variations (SVs) such as large insertions, deletions, inversions, duplications and translocations play a crucial role in disease development, including hereditary cancer. Long-read sequencing, enhanced by QIAseq xHYB Long Read Panels, provides a more complete and accurate view of the cancer genome, facilitating better biomarker discovery and precision medicine applications.
Advantages over amplicon-based targeting
While amplicon-based methods are commonly used for targeted sequencing, they present several limitations, including:
- Primer design challenges – Highly polymorphic and repetitive regions are difficult to amplify efficiently
- Allele dropout – Mismatches in primer binding can lead to incomplete or biased representation of heterozygous regions
- Limited scalability – Multiplexing many large amplicons is problematic as the interrogated genomic regions expand
- Structural variant resolution issues – Breakpoints in large SVs can be poorly defined, making primer placement difficult and biased
In contrast, QIAseq xHYB Long Read Panels provide a hybrid capture-based solution that eliminates these issues, enabling more reliable and unbiased detection of complex genomic variations.
High-fidelity DNA libraries with exceptional uniformity
The QIAseq xHYB Long-Read Kit has been optimized to produce high-quality human DNA libraries with exceptional fidelity. The hybrid capture probe design ensures uniform coverage across high-GC and other traditionally challenging genomic regions, minimizing bias and maximizing sequencing accuracy.
Principle
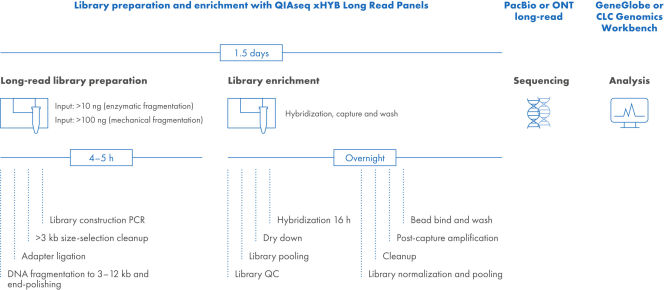
QIAseq xHYB Long-Read Panels are engineered to unlock the full potential of long-read sequencing, delivering unparalleled performance and efficiency. The library preparation workflow generates 3–10 kb DNA fragments through either mechanical (centrifuge-based) or high-throughput enzymatic fragmentation. Both methods employ distinct end-polishing and A-addition steps before adapter ligation. A subsequent size selection removes fragments <3 kb to prevent amplification bias. Finally, PCR indexing enables sample pooling and generates sufficient material for hybrid capture.
The library enrichment leverages QIAseq xHYB hybridization capture strategy for target enrichment from 3–10 kb fragments. Designed for performance, these panels ensure exceptional coverage uniformity, specificity, and sensitivity, empowering researchers to delve deeper into genomic complexities.
Procedure
The QIAseq xHYB Long-Read workflow begins with enzymatic or mechanical fragmentation optimized to produce DNA fragments between 3 kb and 10 kb in length. Mechanical fragmentation is performed with Covaris g-TUBEs using physical DNA shearing based on centrifuge speed. Mechanical shearing is not optimized for high-throughput fragmentation, as tube handling and number are restricted by centrifuge size and physical manipulation of g-TUBEs. In contrast, enzymatic fragmentation facilitates high-throughput fragmentation due to a simple, multi-channel pipette-based workflow. For mechanical fragmentation, the DNA is size-selected after fragmentation to concentrate the fragmented DNA from the g-TUBE fragmentation volume (50 µL) and remove fragmented DNA smaller than 3 kb. The mechanical and enzymatic fragmentation workflows utilize different end-polishing systems: enzymatic fragmentation is performed with FX-based fragmentation and end-polishing, and end-polishing for mechanical fragmentation uses ERA-based polishing and A-addition. After the fragmented DNA is end-polished, an “A” is added to the 3’ end; this product is ready for long-read adapter ligation, where the adapter is added to both ends of the DNA fragments. Following adapter ligation, bead-based size selection removes DNA fragments smaller than 3 kb, which will preferentially amplify over longer DNA fragments if not removed. The libraries are then amplified using PCR-based indexing to allow sample pooling and produce sufficient mass for subsequent hybrid capture.
Hybrid capture targeted enrichment
The size-selected, PCR-indexed long fragment human libraries are pooled with equal mass from each library, and an enhanced blocking buffer is added to prevent non-specific hybridization. The pooled libraries are then dried down using a SpeedVac system. The dried-down pooled libraries are resuspended and denatured. The QIAseq xHYB Long-Read panel is mixed with Hybridization Mix and denatured in a separate tube. After both pooled libraries and the QIAseq xHYB Long-Read panel cool, the xHYB Long-Read panel is added to the pooled libraries, and this is placed overnight in a thermal cycler where the probes will hybridize to their targets. After overnight incubation, the biotinylated probes and any capture products are bound to streptavidin-coated beads. The bound probes and streptavidin beads are washed to remove any unbound non-specific library fragments, and the streptavidin-bound library is resuspended with LR-amp enhancer; elution is performed with sodium hydroxide and neutralized with HN buffer. A small volume pre-amplification is then performed to convert the single-stranded captured DNA into double-stranded libraries and produce sufficient mass to undergo efficient secondary amplification in a larger volume to yield enough material for long-read sequencing library processing for either PacBio or Oxford Nanopore long-read sequencing platforms. A final size selection is performed after amplification, quality-control on the resulting libraries with Qubit BR quantitation and size-distribution assessment using one of the following approaches: 0.7% agarose gel electrophoresis, QIAxcel or TapeStation genomic DNA assay are essential to determine double-stranded DNA concentration and size-distribution of the capture libraries.
Long-read sequencing
For PacBio long-read sequencing, libraries are ready for SMRTbell adapter ligation after performing QC. Please refer to Pacific Biosciences SMRTbell adapter ligation and loading protocols.
For Oxford Nanopore long-read sequencing, refer to Appendix B: Protocol for Oxford Nanopore Sequencing.
Data analysis
The GeneGlobe Data Analysis portal offers simplified downstream data analysis for HLA haplotyping. The QIAGEN CLC Genomics Workbench is used to detect large structural variants in hereditary cancers.
Applications
Repeat expansion disorder analysis
Accurately detect and characterize repeat expansions associated with neurological and genetic disorders, such as Huntington’s disease, Fragile X syndrome and myotonic dystrophy, ensuring precise identification of pathogenic expansions.
HLA typing
Long-read sequencing resolves highly polymorphic HLA regions, enabling accurate HLA genotyping for applications in transplant compatibility, autoimmune disease research and immunogenomics.
Phasing of genetic variants
Determine the haplotype structure of genomic variants over extended distances, providing critical insights into inheritance patterns, allele-specific expression and population genetics.
Structural variant detection
Identify large-scale genomic alterations, including insertions, deletions, inversions, duplications and translocations, with high accuracy, supporting research in cancer genomics, genetic disease studies and evolutionary biology.
Supporting data and figures
QIAseq xHYB long-read library construction and target enrichment workflow
The QIAseq xHYB Long-Read workflow starts with fragmentation of DNA (3–10 kb), using either enzymatic or mechanical methods. Mechanical fragmentation uses Covaris g-TUBEs and is limited in throughput due to centrifuge-based handling. In contrast, enzymatic fragmentation allows high-throughput processing via pipetting. After mechanical fragmentation, DNA is size selected to enrich for ≥3 kb fragments. The two methods also use different end-polishing systems: enzymatic uses FX-based, while mechanical uses ERA-based polishing and A-addition. Fragmented DNA is then A-tailed and ligated with adapters at both ends. A bead-based cleanup removes fragments <3 kb to avoid bias in amplification. Final libraries are PCR-indexed for pooling and hybrid capture.

Resources
Kit Handbooks (1)
Scientific Posters (1)
Brochures & Guides (1)
Safety Data Sheets (1)
Certificates of Analysis (1)