
Get 5% off the enzyme of your choice.
Use the code ENZ2526 at checkout. Offer valid from December 1, 2025, to March 31, 2026. T&Cs* apply.
HotStarTaq DNA Polymerase (250 U)
Cat no. / ID. 203203
250 units HotStarTaq DNA Polymerase, 10x PCR Buffer, 5x Q-Solution, 25 mM MgCl2
Quantity
250 U
1000 U
5000 U
25,000 U
*Offer valid on orders placed from December 1, 2025, to March 31, 2026, or while stocks last. Offer void where prohibited and cannot be combined with any other promotion. Offer is only valid in the following countries: Australia, Austria, Belgium, Canada, Denmark, Finland, France, Germany, Italy, Luxembourg, Netherlands, New Zealand, Norway, Poland, South Africa, Sweden, Switzerland, United Kingdom, United States of America. Freight and delivery costs are not included. It is the customer's responsibility to ensure that acceptance of this offer will not violate any internal policies of the customer's organization and applicable laws and regulations.
The HotStarTaq DNA Polymerase is intended for molecular biology applications. This product is not intended for the diagnosis, prevention, or treatment of a disease.
Get 5% off the enzyme of your choice.
Use the code ENZ2526 at checkout. Offer valid from December 1, 2025, to March 31, 2026. T&Cs* apply.
Features
- Minimal optimization requirements
- High PCR specificity
- Easy handling and room-temperature setup
Product Details
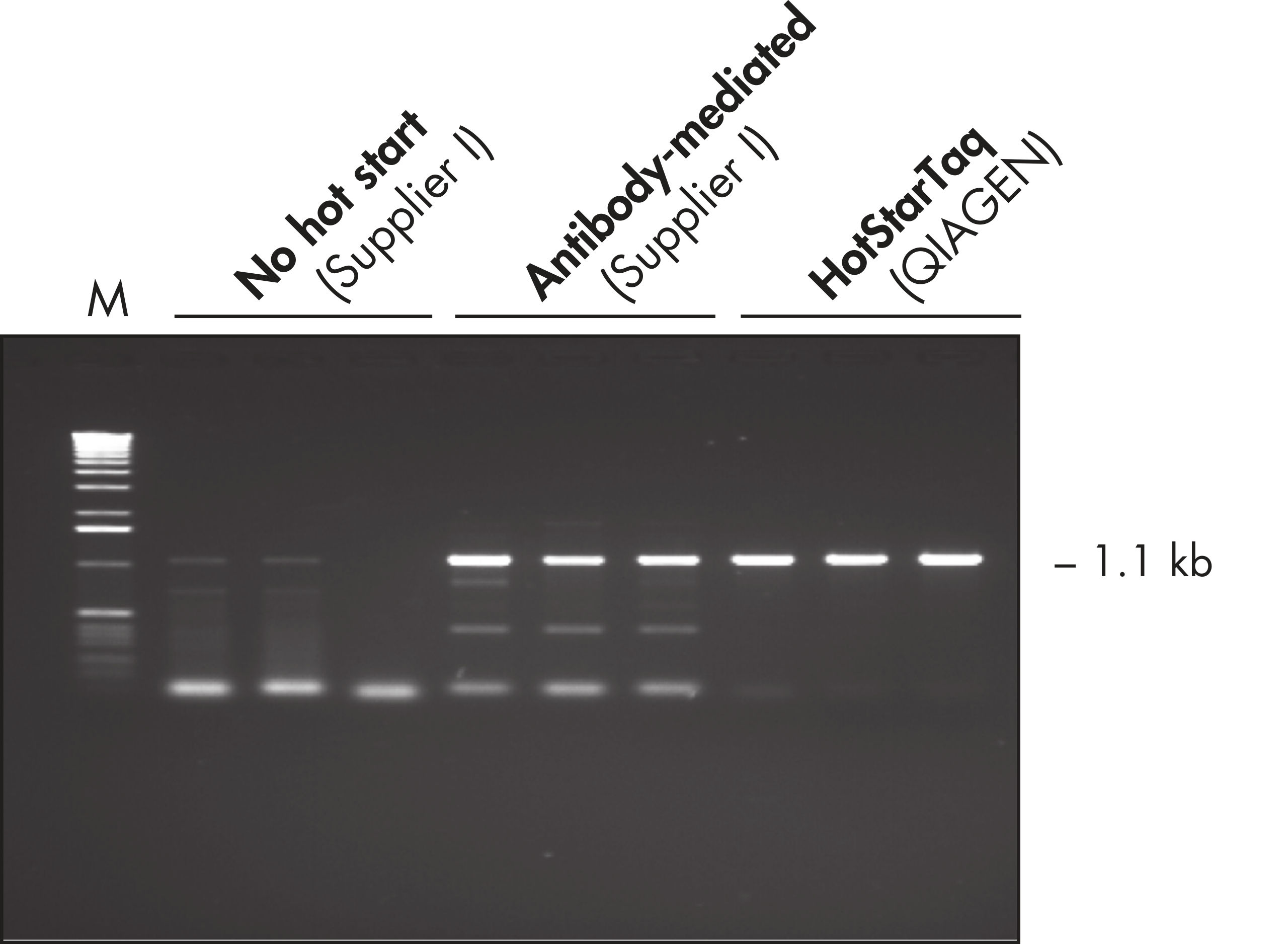
HotStarTaq DNA Polymerase uses a chemically mediated hot start that, unlike, antibody-mediated systems, leads to complete inactivation of the polymerase until the initial heat activation step at the start of PCR. HotStarTaq DNA Polymerase is supplied with the unique QIAGEN PCR Buffer, which minimizes nonspecific amplification products, primer dimers, and background. Q-Solution, a novel additive that enables efficient amplification of "difficult" (e.g., GC rich) templates, is also provided.
Performance
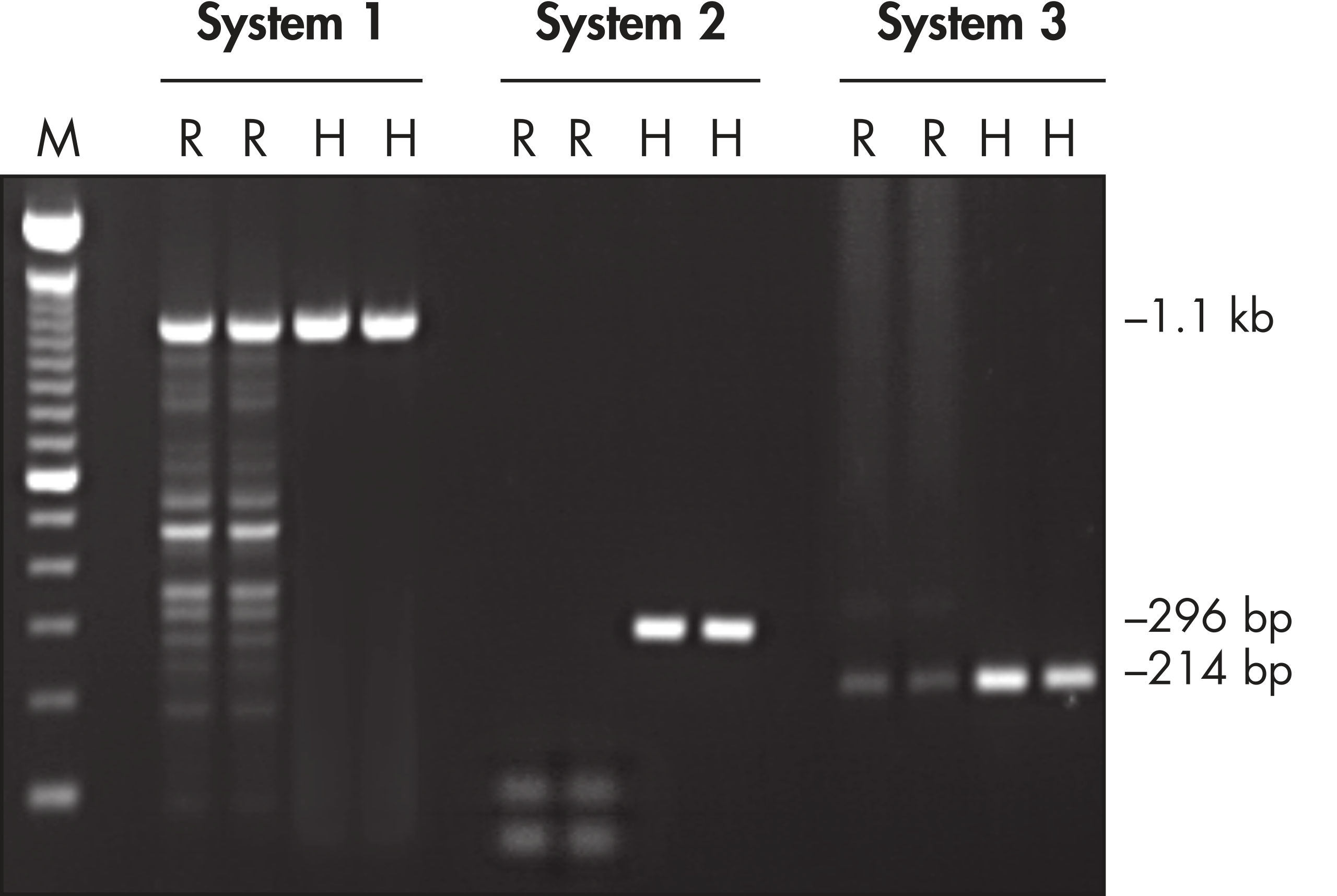
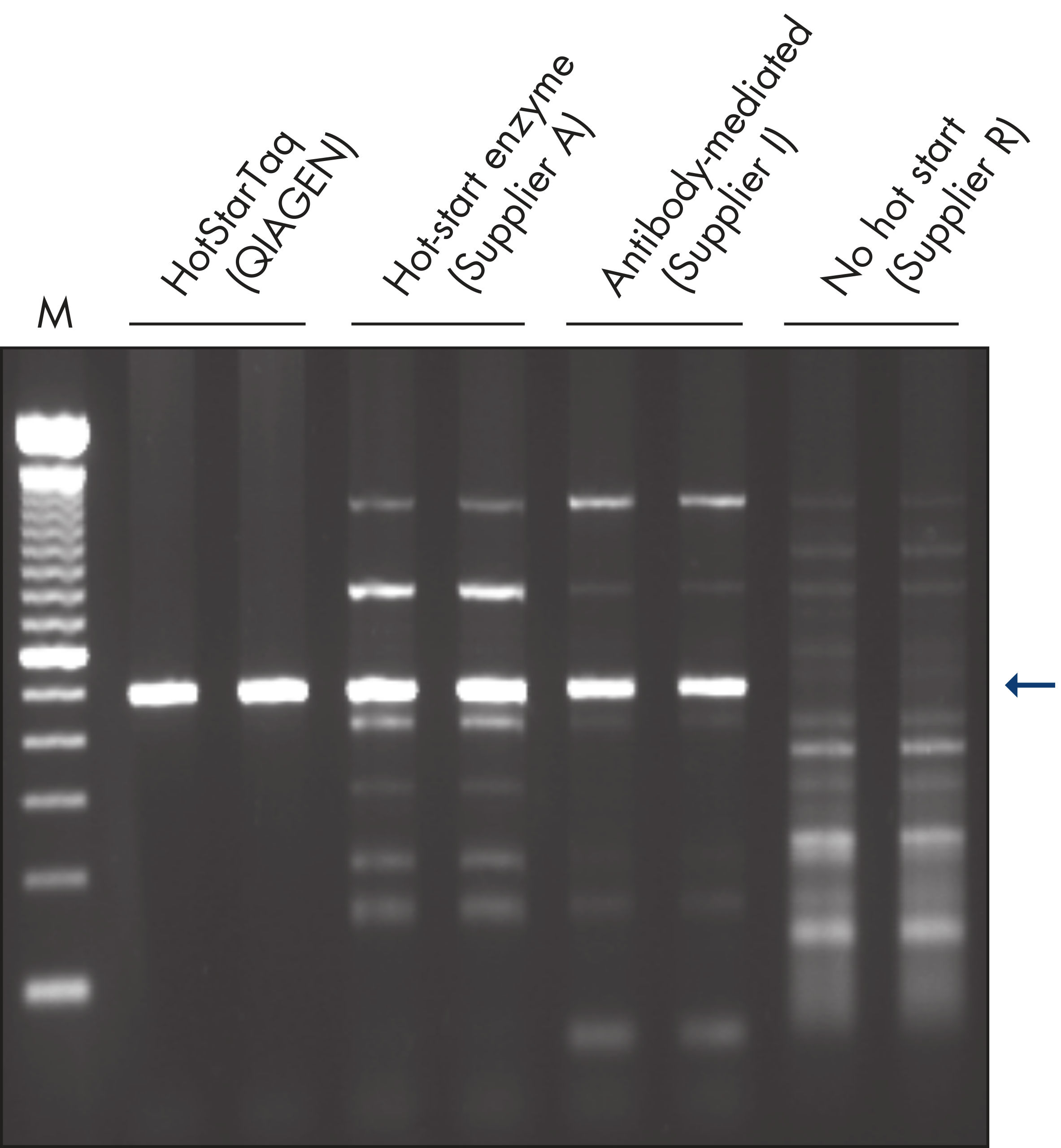
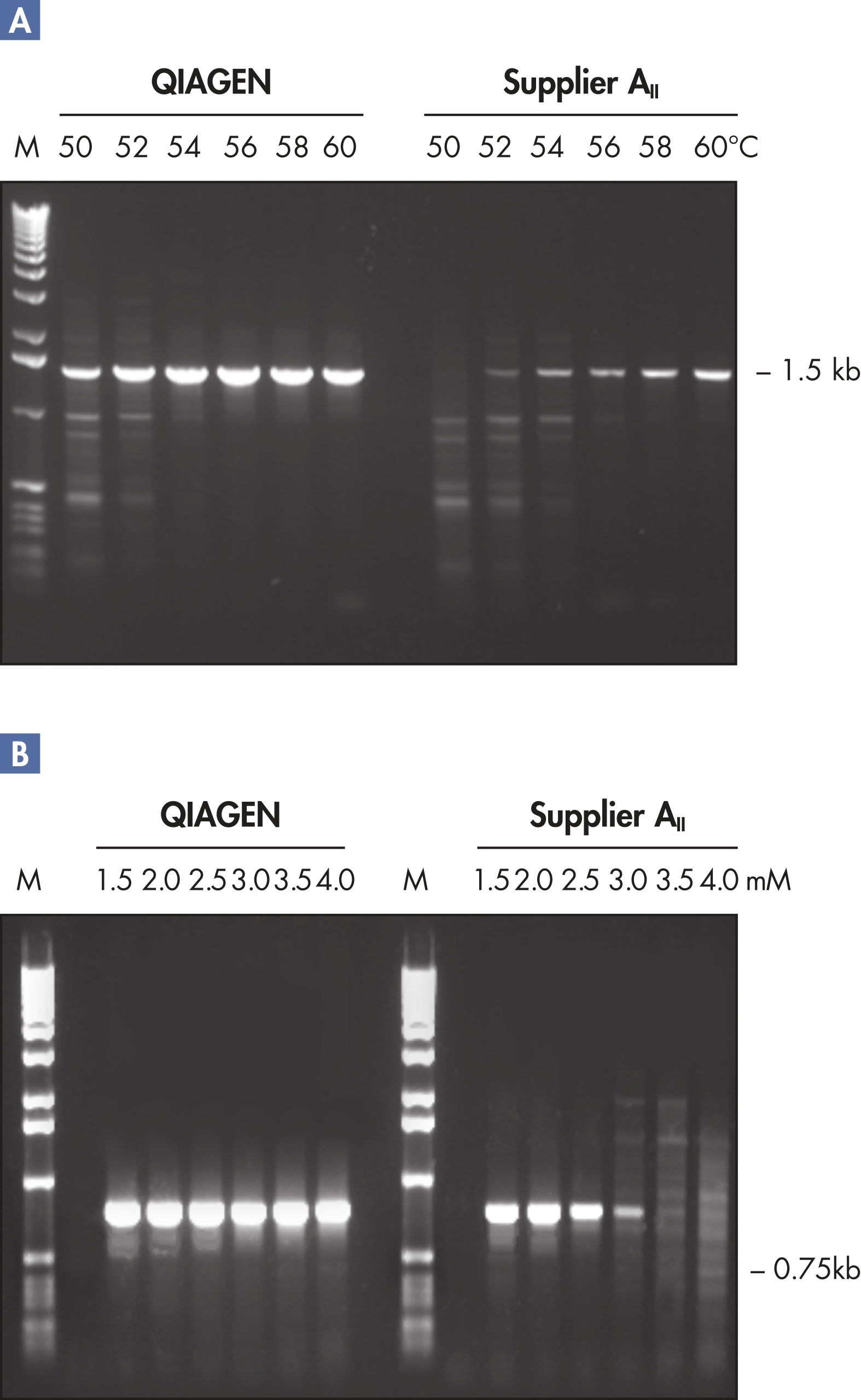
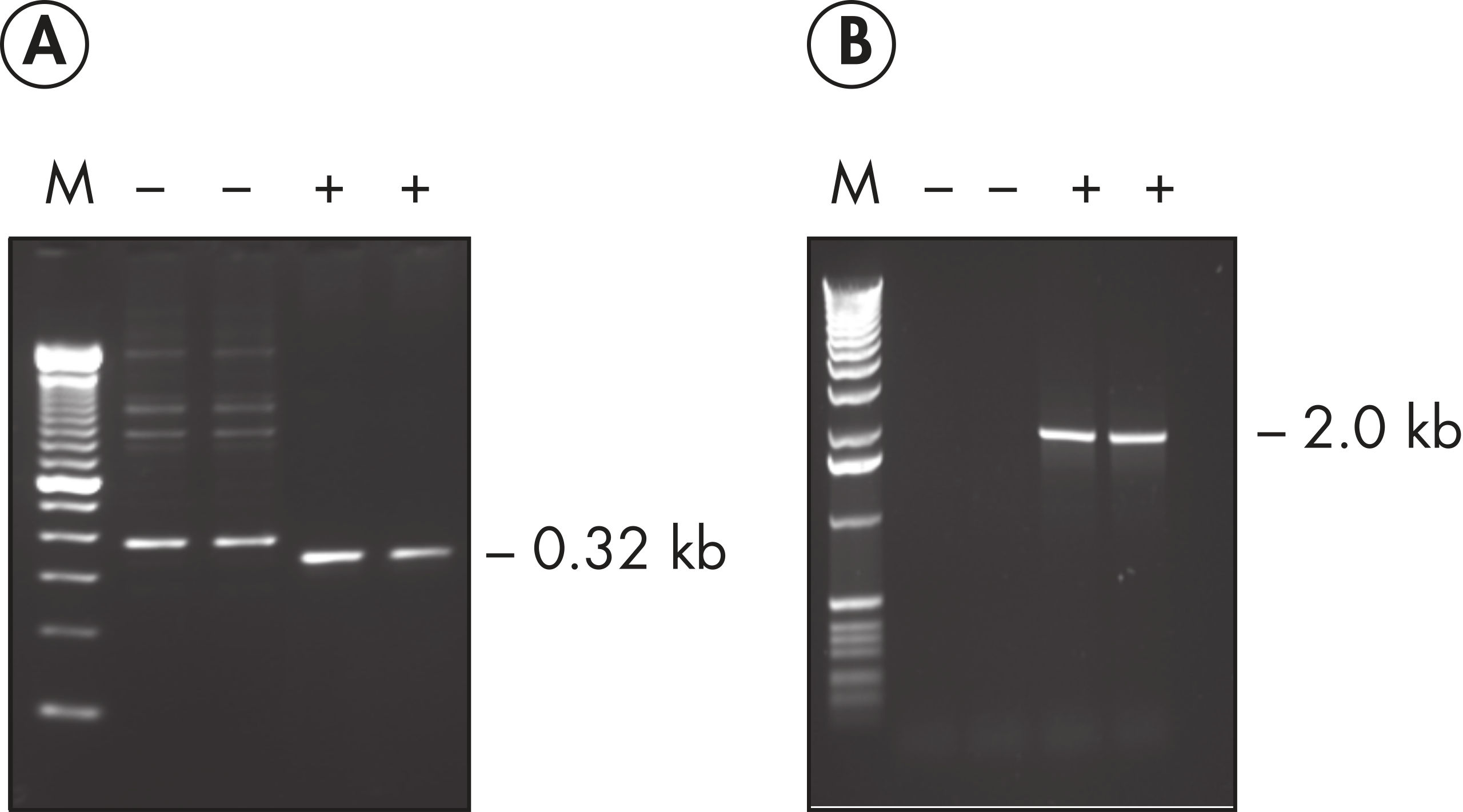
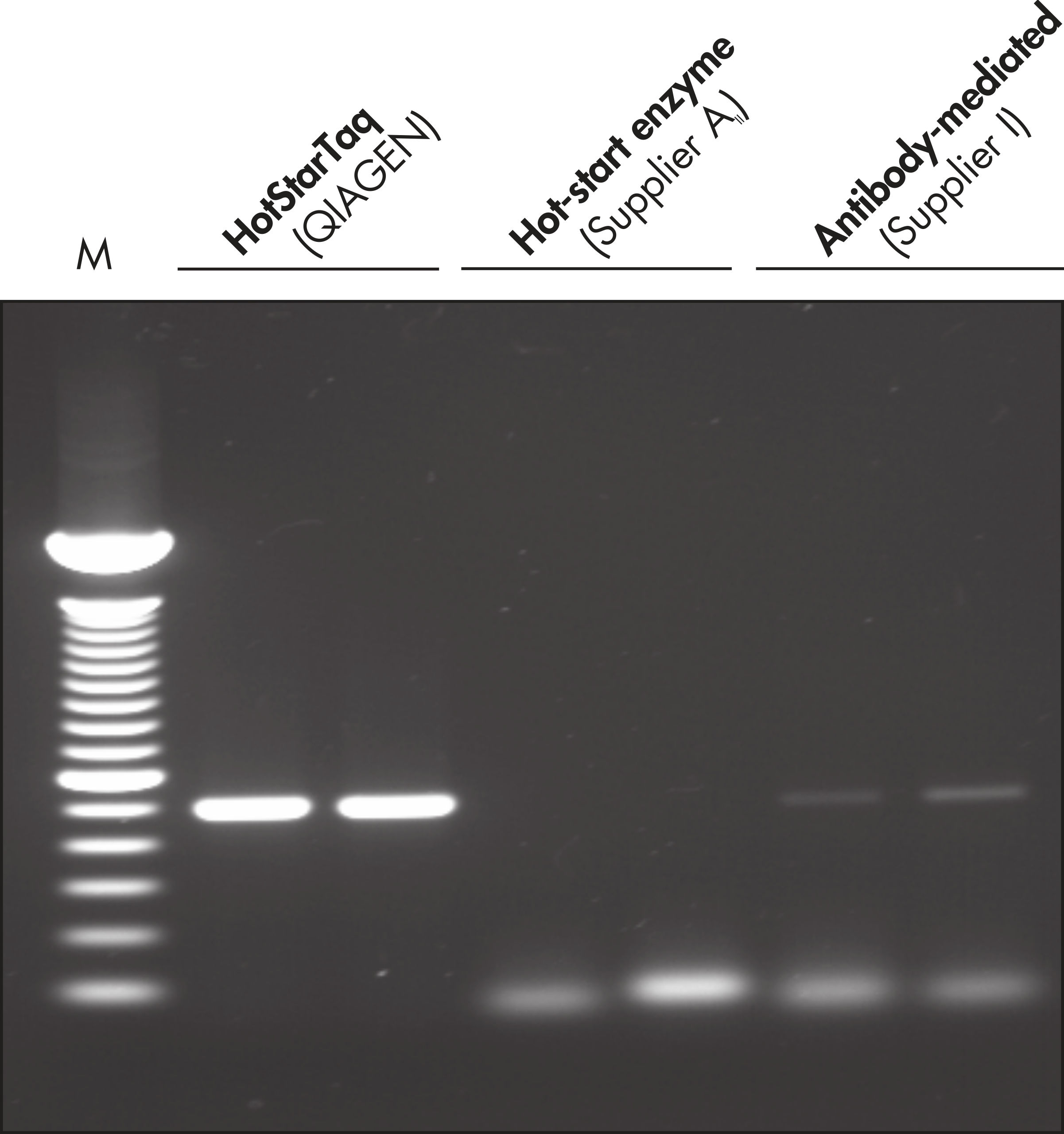
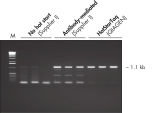
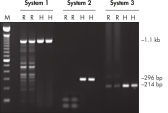
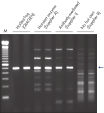
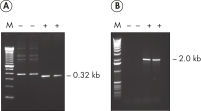
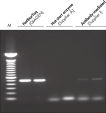
Each lot of HotStarTaq DNA Polymerase is subjected to a comprehensive range of quality control tests, including a stringent PCR specificity and reproducibility assay in which low-copy targets are amplified. HotStarTaq DNA Polymerase outperformed kits tested from other suppliers and ensures high specificity and superior performance in hot-start PCR (see figures " Higher specificity with different primer–template systems" and " Superior performance" and table). The innovative PCR buffer provided with the kit ensures specificity over a wide range of PCR conditions, minimizing the need for optimization (see figure Tolerance to variable temperature and magnesium concentrations). Suboptimal PCR can be improved with Q-Solution, also provided with the kit (see figure " Amplification of difficult templates"). Together, these components ensure specific amplification in a range of applications (see figure " Effect of hot start on RT-PCR performance" and " Highly sensitive single-cell PCR").
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HotStarTaq DNA Polymerase | Hot-start enzyme from Supplier AII | Antibody-mediated | Manual | Wax barrier | |
|---|---|---|---|---|---|
| Specific amplification | ++ | + | + | +/– | +/– |
| Minimal PCR optimization | ++ | +/– | +/– | – | – |
| Easy to use | ++ | ++ | + | – | – |
HotStarTaq DNA Polymerase specifications
Concentration: 5 units/µl
Recombinant enzyme: Yes
Substrate analogs: dNTP, ddNTP, dUTP, biotin-11-dUTP, DIG-11-dUTP, fluorescent-dNTP/ddNTP
Extension rate: 2–4 kb/min at 72°C
Half-life: 10 min at 97°C ; 60 min at 94°C
Amplification efficiency: ≥105 fold
5'–>3' exonuclease activity: Yes
Extra A addition: Yes
3'–>5' exonuclease activity: No
Contaminating nucleases: No
Contaminating RNases: No
Contaminating proteases: No
Self-priming activity: No
{kind=link}
{kind=link}
Principle
HotStarTaq DNA Polymerase, a modified form of Taq DNA Polymerase, provides high specificity in hot-start PCR. The kit includes an innovative dual-cation PCR buffer, Q-Solution, and MgCl2.
HotStarTaq DNA Polymerase
HotStarTaq DNA Polymerase is supplied in an inactive state and has no polymerase activity at ambient temperatures. This prevents extension of nonspecifically annealed primers and primer dimers formed at low temperatures during PCR setup and the initial PCR cycle (see figures " Superior performance in hot-start PCR" and " Higher specificity with different primer–template systems"). HotStarTaq DNA Polymerase is activated by a 15-minute incubation at 95°C, which can be incorporated into any existing thermal-cycler program.
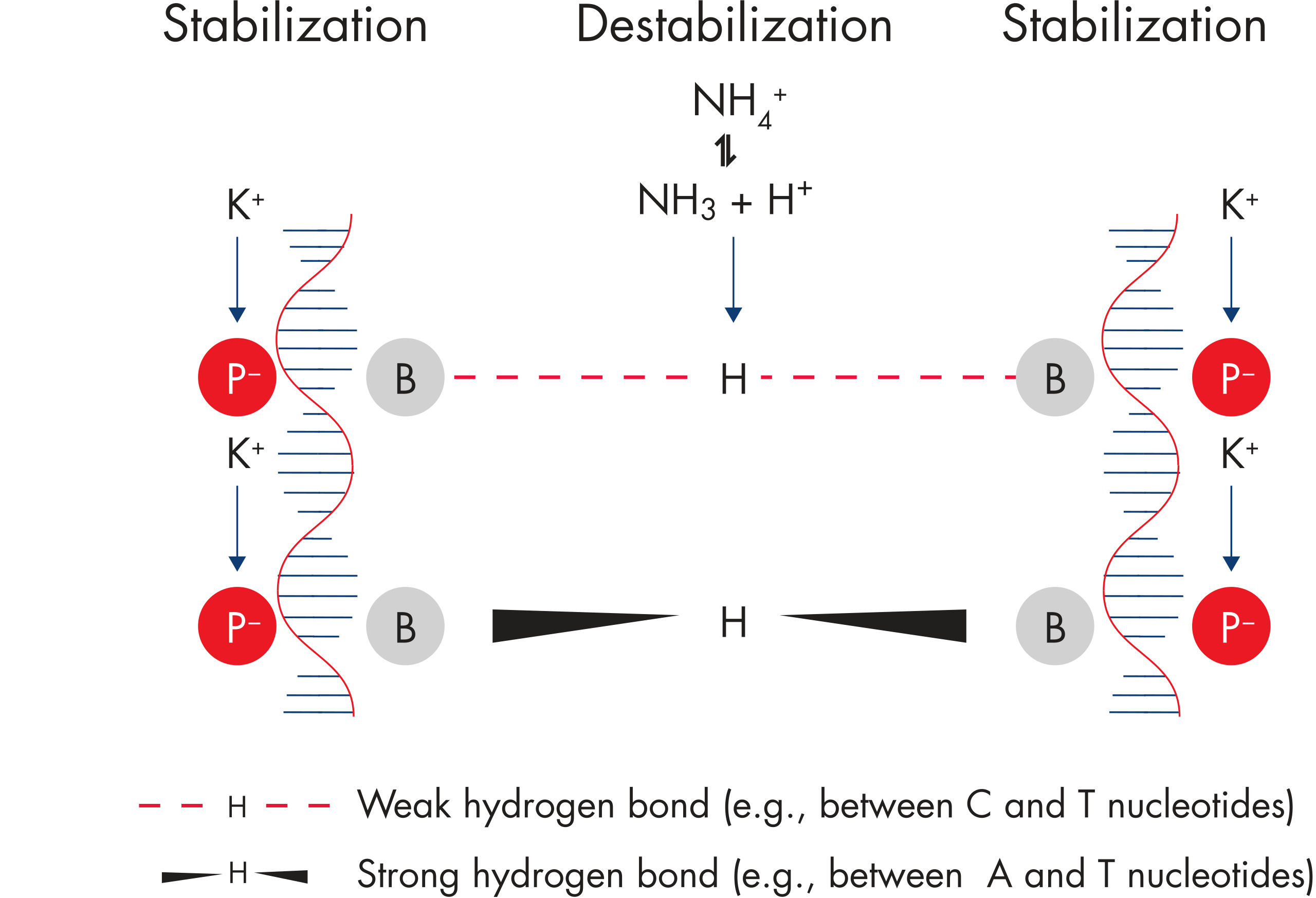
QIAGEN PCR Buffer
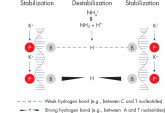
QIAGEN PCR Buffer maintains specific amplification in every cycle of PCR by promoting a high ratio of specific-to-nonspecific primer binding during the annealing step in each PCR cycle (see figure " Increased specificity of primer annealing"). Owing to a uniquely balanced combination of KCl and (NH4)2SO4, the buffer provides stringent primer-annealing conditions over a wider range of annealing temperatures and Mg2+ concentrations than conventional PCR buffers. Optimization of PCR by varying the annealing temperature or the Mg2+ concentration is therefore often minimal or not required (see figure Tolerance to variable temperature and magnesium concentrations).
Q-Solution
Q-Solution, an innovative PCR additive that facilitates amplification of difficult templates by modifying the melting behavior of DNA, is also provided with HotStarTaq DNA Polymerase. This unique reagent improves suboptimal PCR caused by templates that have a high degree of secondary structure or with GC-rich templates (see figure " Amplification of difficult templates"). Unlike other commonly used PCR additives such as DMSO, Q-Solution is used at just one working concentration, is nontoxic, and PCR purity is guaranteed. Adding Q-Solution to the PCR does not compromise PCR fidelity.
Procedure
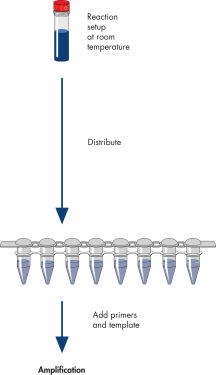
HotStarTaq DNA Polymerase is supplied with a streamlined, optimized protocol for fast and easy PCR setup. HotStarTaq DNA Polymerase is activated by a 15-minute, 95°C incubation step, which can easily be incorporated into existing thermal cycling programs. Reactions can be set up at room temperature, ensuring greater convenience and ease of use (see figure " HotStarTaq procedure").
Applications
HotStarTaq DNA Polymerase is suitable for a wide variety of applications, including challenging applications, such as amplification of:
- Complex genomic templates
- Complex cDNA templates (e.g., RT-PCR)
- Very low-copy targets (e.g., single-cell PCR)
- Reactions with multiple primer pairs
Supporting data and figures
HotStarTaq procedure.
HotStarTaq procedure.

Specifications
| Features | Specifications |
|---|---|
| Applications | PCR, RT-PCR, Complex genomic templates, very low-copy targets |
| With/without hotstart | With hotstart |
| Reaction type | PCR amplification |
| Sample/target type | Genomic DNA and cDNA |
| Real-time or endpoint | Endpoint |
| Enzyme activity | 5' -> 3' exonuclease activity |
| Mastermix | No |
| Single or multiplex | Single |
Resources
Brochures & Guides (5)
Quick-Start Protocols (1)
Kit Handbooks (1)
Supplementary Protocols (2)
Safety Data Sheets (1)
Technical Information (1)
Certificates of Analysis (1)
Publications
MALDI-TOF mass spectrometry for multiplex genotyping of CYP2B6 single-nucleotide polymorphisms.
Clin Chem; 2006; 53 (1):24-33 2006 Nov 2 PMID:17082249
Age-related urinary excretion of BK polyomavirus by nonimmunocompromised individuals.
J Clin Microbiol; 2006; 45 (1):193-8 2006 Nov 8 PMID:17093017
Transcriptional regulation of human CYP2A13 expression in the respiratory tract by CCAAT/enhancer binding protein and epigenetic modulation.
Mol Pharmacol; 2006; 71 (3):807-16 2006 Dec 5 PMID:17148654
Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy.
Proc Natl Acad Sci U S A; 2006; 103 (50):19152-7 2006 Dec 4 PMID:17146052
Assessing combined methylation-sensitive high resolution melting and pyrosequencing for the analysis of heterogeneous DNA methylation.
Epigenetics; 2011; 6 (4):500-7 2011 Apr 1 PMID:21364322
FAQ
What kind of PCR products can be cloned with the QIAGEN PCR Cloning Kit?
Do CoralLoad dyes supplied in various QIAGEN PCR Kits interfere with downstream applications?
Does QIAGEN sell Q-Solution separately?
How can one determine the optimal annealing temperature for a specific PCR assay?
Do any of the buffers in the HotStarTaq DNA Polymerase Kit contain Triton?
Is Q-Solution required for PCR with QIAGEN's PCR kits?
Can Taq DNA Polymerase use RNA as a template, and generate false positives in "no-RT" controls?
How can I avoid primer-dimer formation during PCR amplification?
How can I tell if I have primer-dimers in my PCR reaction?
Can I shorten the activation time for the HotStarTaq DNA Polymerase?
What makes QIAGEN's 10x Taq and HotStarTaq DNA Polymerase PCR buffer superior?
What is the composition of the QIAGEN 10x PCR Buffer in Taq- and HotStarTaq DNA Polymerase Kits?
What should the starting template DNA quality and quantity be for PCR?
Can QIAGEN's Taq- and HotstarTaq DNA Polymerases be used for cycle sequencing?
How is "Touchdown PCR" used to increase PCR specificity?
How much DNA is obtained in the average PCR reaction?
Have you tested the effect of inhibitors on PCR performance?
Why do I get smeared PCR products?
Do you have a protocol for polyacrylamide gel analysis of oligonucleotides?