
Need bulk, customized or optimized products for commercial purposes? We also offer support with logistics, compliance and more. Reach out to cooperate with QIAGEN Strategic Partnerships & OEM
Taq DNA Polymerase (250 U)
Cat. No. / ID: 201203
250 units Taq DNA Polymerase, 10x PCR Buffer, 10x CoralLoad PCR Buffer, 5x Q-Solution, 25 mM MgCl2
Log in To see your account pricing.
Quantity
250 U
1000 U
5000 U
25,000 U
The Taq DNA Polymerase is intended for molecular biology applications. This product is not intended for the diagnosis, prevention, or treatment of a disease.
Need bulk, customized or optimized products for commercial purposes? We also offer support with logistics, compliance and more. Reach out to cooperate with QIAGEN Strategic Partnerships & OEM
Features
- QIAGEN PCR Buffer for minimal optimization
- Additional ready-to-load PCR buffer for faster handling
- Q-Solution for amplification of GC-rich templates
- Choice of formats for convenience and ease of handling
Product Details
Taq DNA Polymerase is supplied with the unique QIAGEN PCR Buffer that minimizes the need for optimization of PCR parameters, as well as Q-Solution, a novel additive that enables efficient amplification of "difficult" (e.g., GC rich) templates. In addition, CoralLoad PCR Buffer (containing two gel-tracking dyes) is also provided, enabling immediate loading of PCR products.
Performance
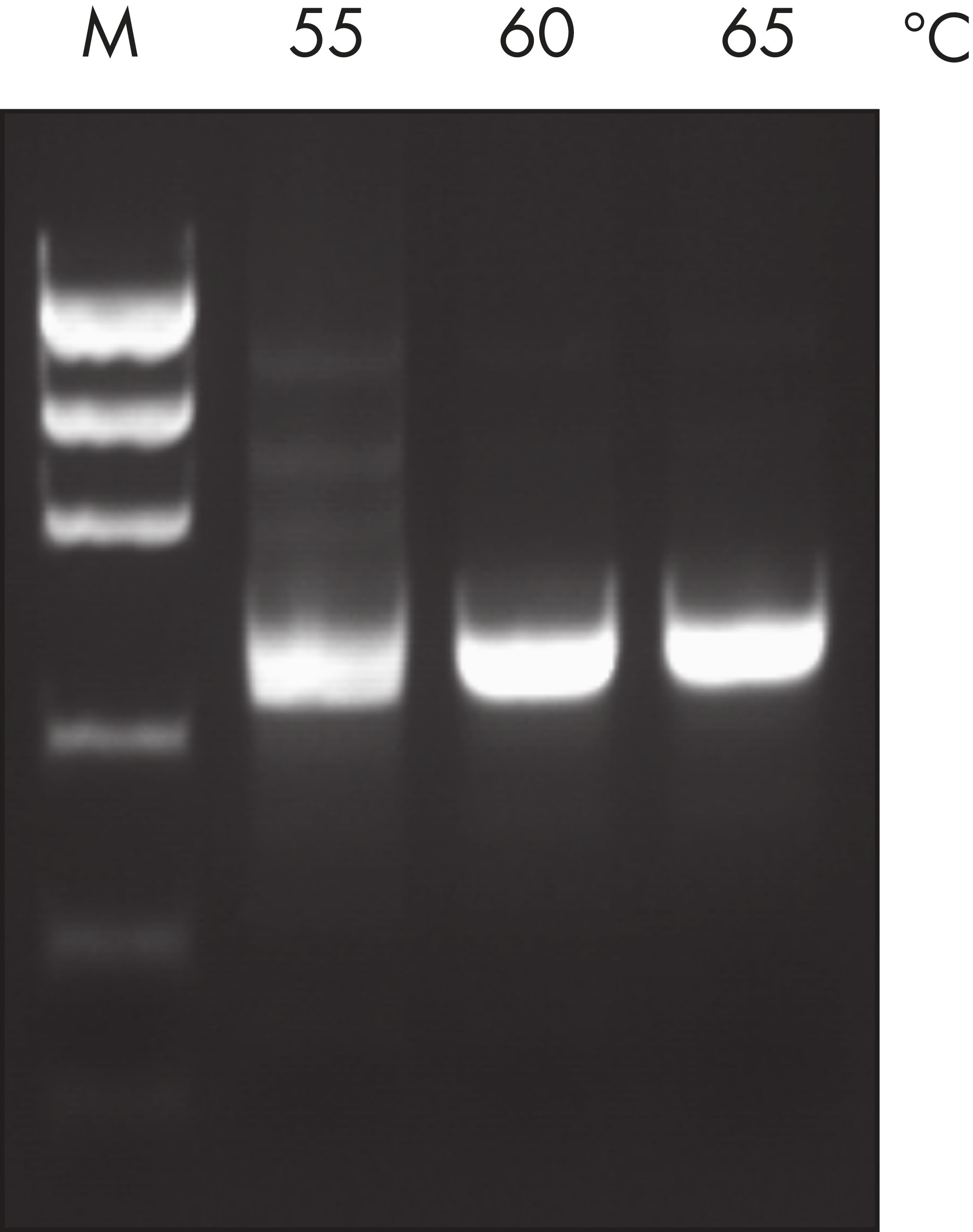
Taq DNA Polymerase outperformed kits tested from other suppliers and delivers robust PCR performance in a wide range of PCR conditions, without the need for time-consuming optimization (see figures " Tolerance of different primer Tm Values" and " Specific amplification of long PCR products"). Every lot of Taq DNA Polymerase is subjected to a comprehensive range of quality control tests, including a stringent PCR specificity and reproducibility assay in which low-copy targets are amplified from human genomic DNA (see figure " Lot-to-lot reproducibility"). The unique formulation of QIAGEN PCR Buffer and CoralLoad PCR Buffer, also provided with the kit, enable highly specific PCR in a variety of PCR conditions with minimal optimization requirements (see figure " Wide annealing-temperature window and Tolerance to variable magnesium concentration"). In addition, CoralLoad PCR Buffer enables immediate loading of PCR products onto an agarose gel for even easier handling and faster results. Suboptimal PCR can be improved using Q-Solution, a PCR additive, also provided with the kit (see figure " Amplification of difficult templates").
Taq DNA Polymerase specifications
Concentration: 5 units/µl
Recombinant enzyme: Yes
Substrate analogs: dNTP, ddNTP, dUTP, biotin-11-dUTP, DIG-11-dUTP, fluorescent-dNTP/ddNTP
Extension rate: 2–4 kb/min at 72°C
Half-life: 10 min at 97°C; 60 min at 94°C
Amplification efficiency: ≥105 fold
5'–>3' exonuclease activity: Yes
Extra A addition: Yes
3'–>5' exonuclease activity: No
Contaminating nucleases: No
Contaminating RNases: No
Contaminating proteases: No
Self-priming activity: No >
See figures
Principle
Taq DNA Polymerase is a high-quality recombinant enzyme that is suitable for general and specialized PCR applications (see figures " Tolerance of different primer Tm Values" and " Specific amplification of long PCR products").
QIAGEN PCR Buffer
Innovative QIAGEN PCR Buffer has been developed to save time and effort by reducing the need for PCR optimization. QIAGEN PCR Buffer contains both KCl and (NH4)2SO4(see figure " Increased specificity of primer annealing"). This unique buffer facilitates the amplification of specific PCR products. During the annealing step of every PCR cycle, the buffer allows a high ratio of specific-to-nonspecific primer binding. Owing to a uniquely balanced combination of KCl and (NH4)2SO4, the PCR buffer provides stringent primer-annealing conditions over a wider range of annealing temperatures and Mg2+ concentrations than conventional PCR buffers. Optimization of PCR by varying the annealing temperature or the Mg2+ concentration is dramatically reduced and often not required (see figure " Wide annealing temperature window and Tolerance of variable magnesium concentration").
CoralLoad PCR Buffer
CoralLoad PCR Buffer has all the advantages of QIAGEN PCR Buffer. In addition, it can also be used to directly load the PCR reaction onto an agarose gel — separate addition of a gel loading buffer is not required. CoralLoad PCR Buffer provides the same high PCR specificity and minimal reaction optimization as the conventional QIAGEN PCR Buffer. Additionally, it contains two marker dyes — an orange dye and a red dye — that facilitate estimation of DNA migration distance and optimization of agarose gel run time (see figure " CoralLoad PCR Buffer"). The buffer ensures improved pipetting visibility and enables direct loading of PCR products onto a gel, for enhanced convenience.
Q-Solution
Q-Solution facilitates amplification of GC-rich templates or templates with a high degree of secondary structure by modifying the melting behavior of DNA. Use of this unique reagent often enables or improves suboptimal PCR (see figure " Amplification of difficult templates"). Unlike DMSO and other PCR additives, Q-Solution is used at a defined working concentration with any primer–template system and is not toxic.
See figures
Tolerance of different primer Tm values.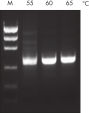 Specific amplification of long PCR products.
Specific amplification of long PCR products.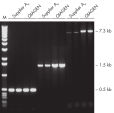 NH4+ and K+ cations in QIAGEN PCR buffers increase specific primer annealing.
NH4+ and K+ cations in QIAGEN PCR buffers increase specific primer annealing.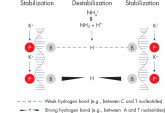 A. Wide annealing temperature window. B. Tolerance of variable magnesium concentration.
A. Wide annealing temperature window. B. Tolerance of variable magnesium concentration. CoralLoad PCR Buffer.
CoralLoad PCR Buffer.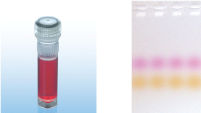 Amplification of difficult templates with Q-Solution.
Amplification of difficult templates with Q-Solution.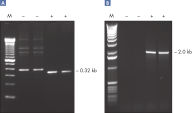
 Specific amplification of long PCR products.
Specific amplification of long PCR products.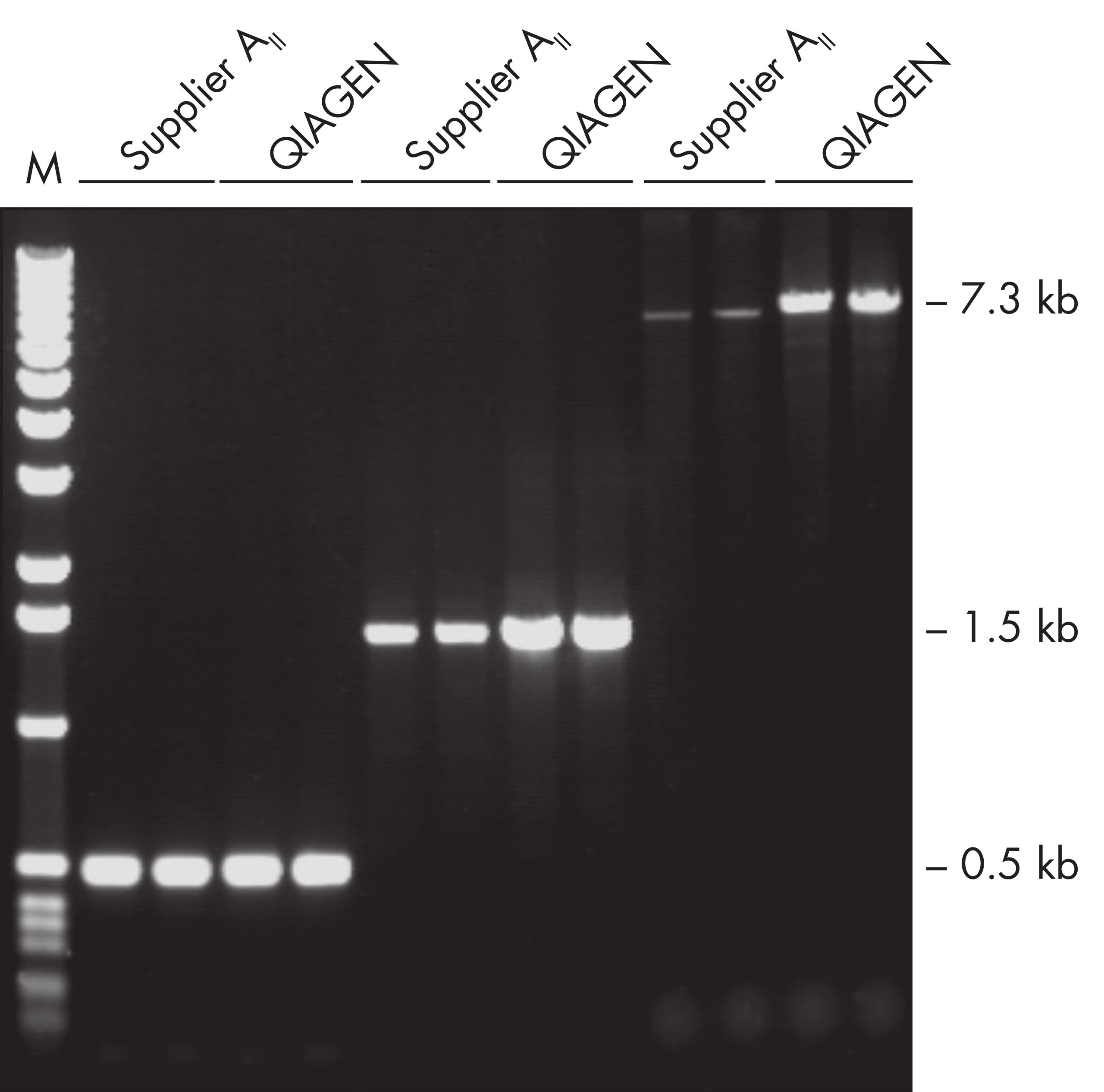 NH4+ and K+ cations in QIAGEN PCR buffers increase specific primer annealing.
NH4+ and K+ cations in QIAGEN PCR buffers increase specific primer annealing.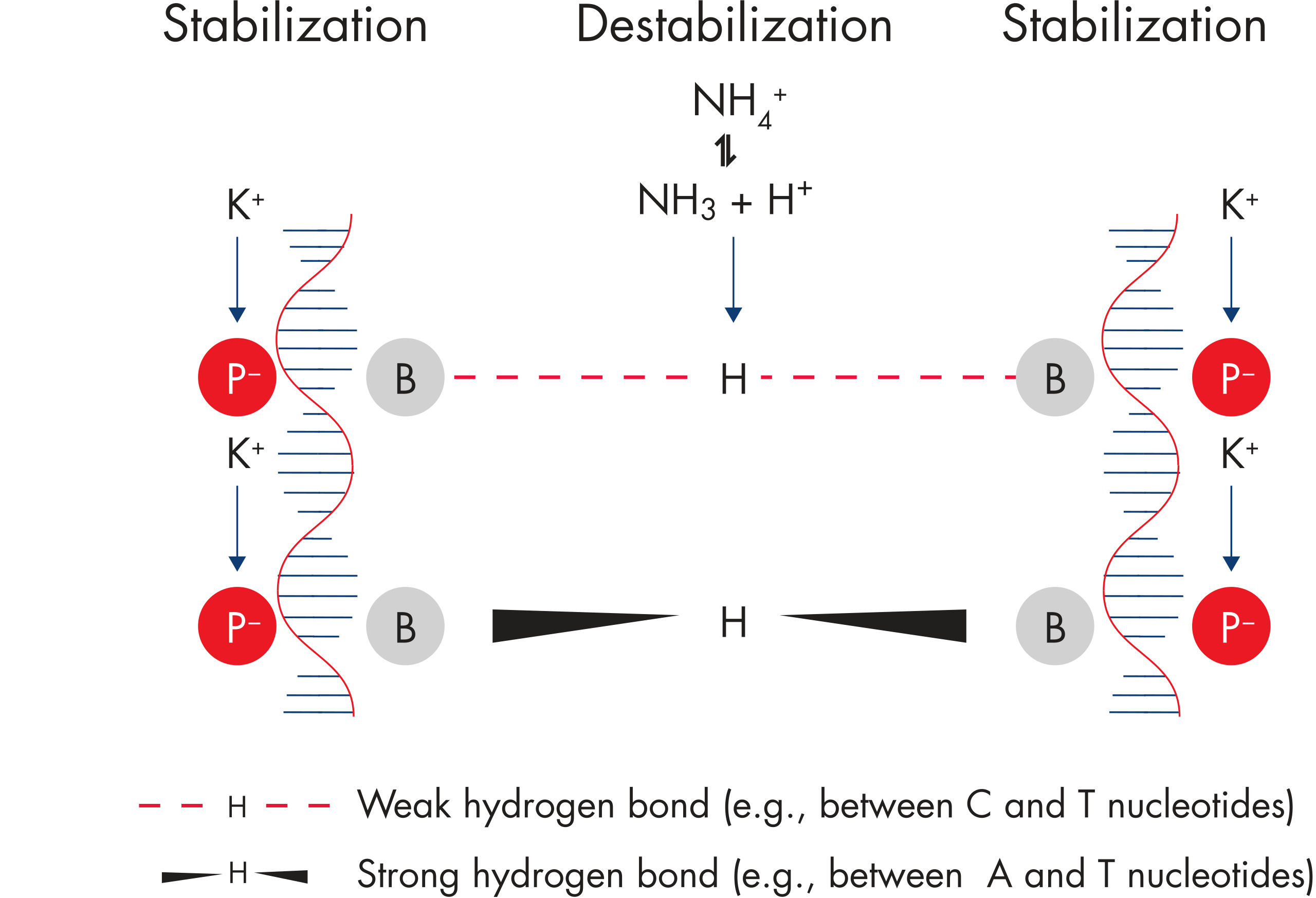 A. Wide annealing temperature window. B. Tolerance of variable magnesium concentration.
A. Wide annealing temperature window. B. Tolerance of variable magnesium concentration.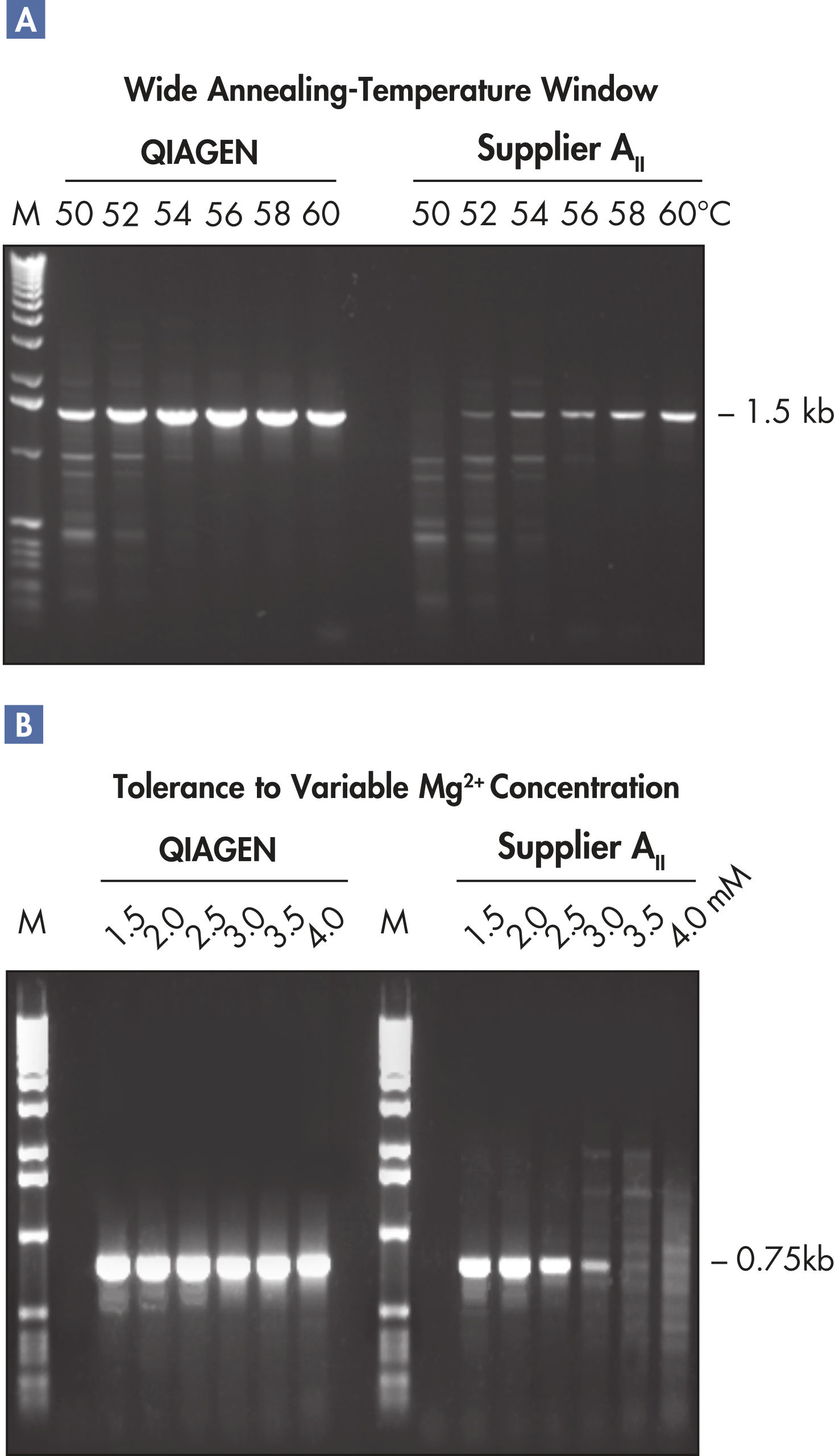 CoralLoad PCR Buffer.
CoralLoad PCR Buffer.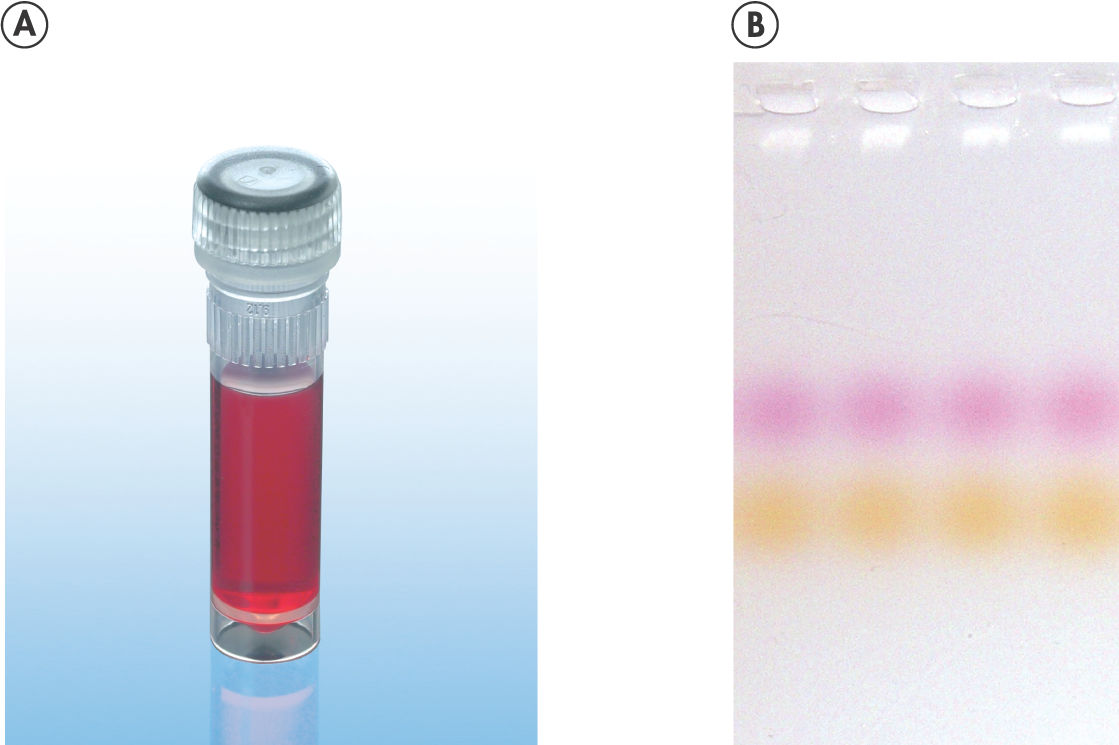 Amplification of difficult templates with Q-Solution.
Amplification of difficult templates with Q-Solution.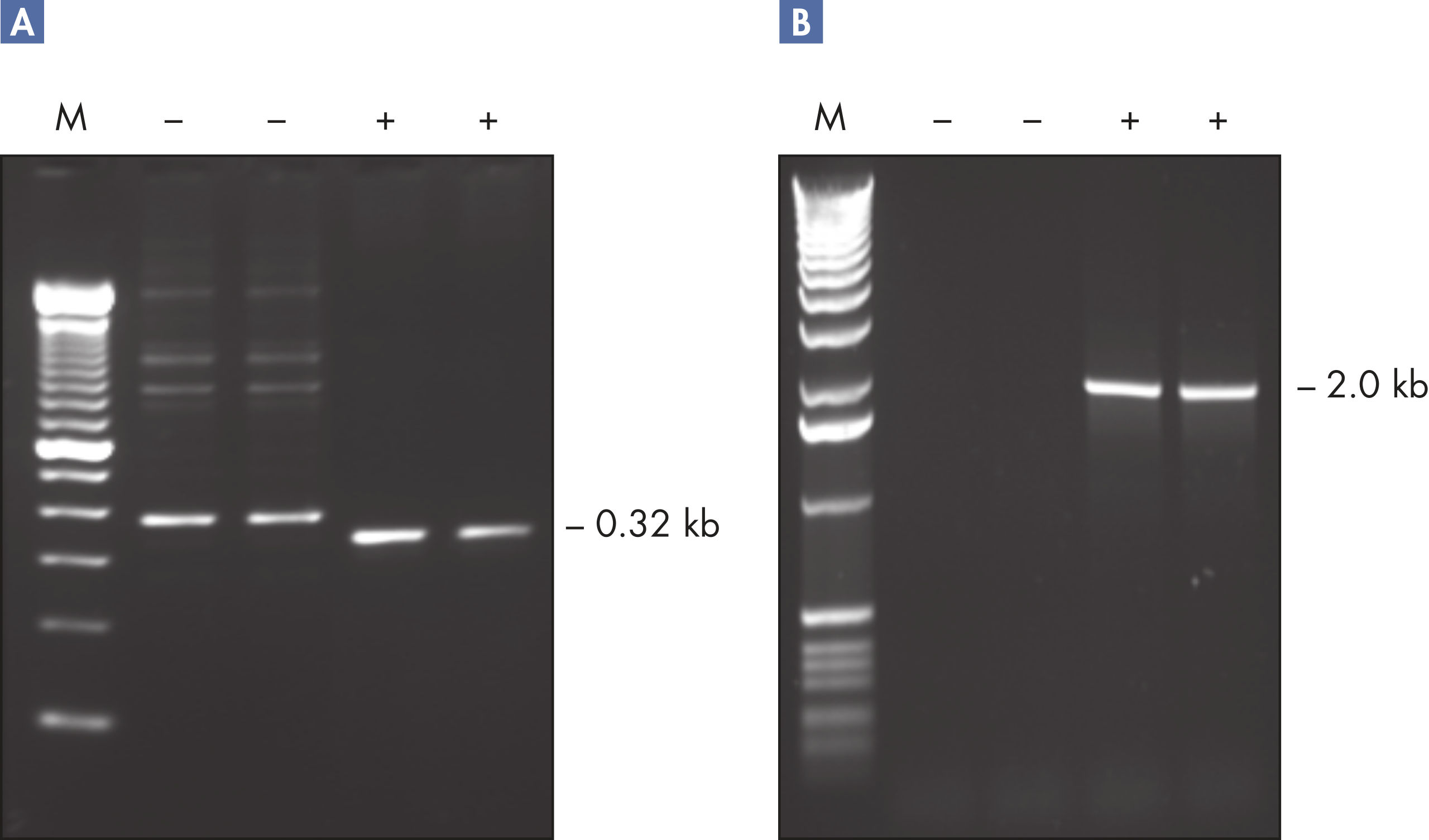
Procedure
Taq DNA Polymerase ensures highly specific PCR for a range of applications — with minimal optimization of PCR parameters. The streamlined, easy-to-follow protocol provided with the kit simplifies PCR setup. For added convenience and easier handling, CoralLoad PCR Buffer is provided. PCR products can be directly loaded onto a gel without the addition of a loading dye. To ensure success with GC-rich templates, the PCR enhancer Q-Solution is included.
Applications
Taq DNA Polymerase is used for standard and specialized applications, including:
- General PCR
- RT-PCR
- Screening
- PCR-based DNA fingerprinting (VNTR, STR, and RAPD)
Supporting data and figures
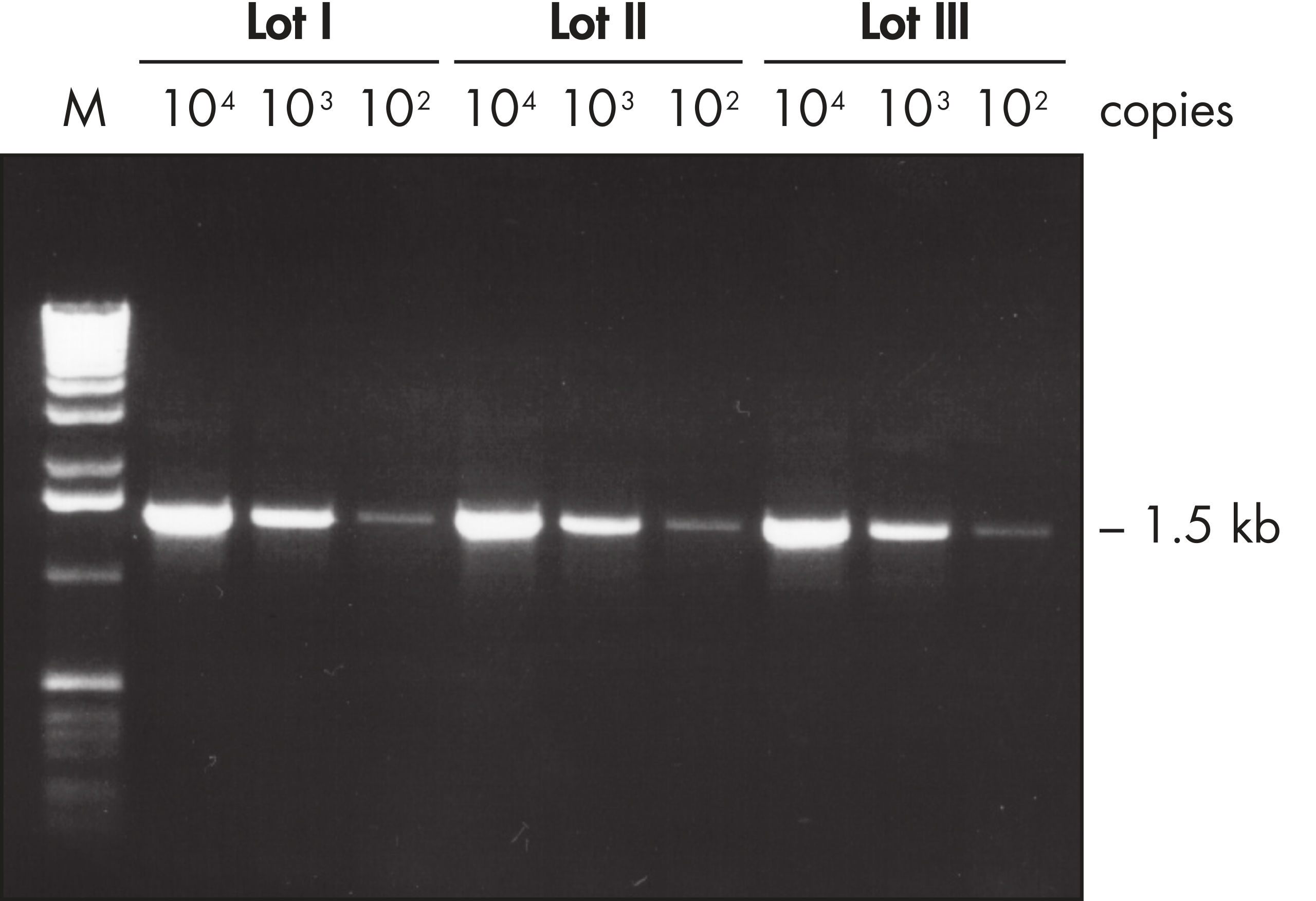
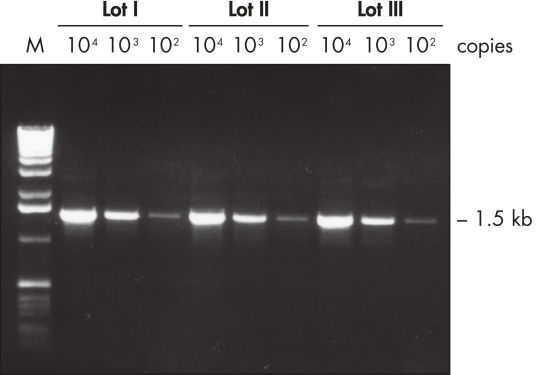
Lot-to-lot reproducibility.
A fragment of the single-copy gene for cystic fibrosis was amplified from 30 ng, 3 ng, and 300 pg human genomic DNA corresponding to 104, 103, and 102 copies of target template, respectively. Three different lots of QIAGEN Taq DNA Polymerase were used and equal volumes of the PCR product were analyzed on a 1% agarose gel. M: markers.
Specifications
| Features | Specifications |
|---|---|
| Applications | PCR, RT-PCR, DNA fingerprinting |
| dNTP's included | No |
| Real-time or endpoint | Endpoint |
| Reaction type | PCR amplification |
| Single or multiplex | Single |
| With/without hotstart | Without hotstart |
| Enzyme activity | 5' -> 3' exonuclease activity |
| Mastermix | No |
| Sample/target type | Genomic DNA and cDNA |
Resources
Brochures & Guides (3)
Quick-Start Protocols (1)
Safety Data Sheets (1)
Supplementary Protocols (1)
Kit Handbooks (1)
Technical Information (1)
Certificates of Analysis (1)
Publications
Increased expression of matrix metalloproteinase-9 in the eutopic endometrial tissue of women with endometriosis.
Hum Reprod; 2006; 21 (12):3059-67 2006 Jul 31 PMID:16880228
Kaposi sarcoma herpesvirus-encoded vFLIP and vIRF1 regulate antigen presentation in lymphatic endothelial cells.
Blood; 2006; 109 (4):1550-8 2006 Oct 17 PMID:17047149
Progesterone receptor polymorphism +331G/A is associated with a decreased risk of deep infiltrating endometriosis.
Hum Reprod; 2006; 22 (1):129-35 2006 Aug 18 PMID:16920727
Haplotype analysis of the DQA genes in sheep: evidence supporting recombination between the loci.
J Anim Sci; 2006; 85 (3):577-82 2006 Nov 22 PMID:17121973
Role of RelGsu in stress response and Fe(III) reduction in Geobacter sulfurreducens.
J Bacteriol; 2006; 188 (24):8469-78 2006 Oct 13 PMID:17041036
FAQ
How is "Touchdown PCR" used to increase PCR specificity?
What should the starting template DNA quality and quantity be for PCR?
What kind of PCR products can be cloned with the QIAGEN PCR Cloning Kit?
Why do I get smeared PCR products?
Does QIAGEN sell Q-Solution separately?
Do CoralLoad dyes supplied in various QIAGEN PCR Kits interfere with downstream applications?
Can Taq DNA Polymerase use RNA as a template, and generate false positives in "no-RT" controls?
Is Q-Solution required for PCR with QIAGEN's PCR kits?
How can I avoid primer-dimer formation during PCR amplification?
How can I tell if I have primer-dimers in my PCR reaction?
What makes QIAGEN's 10x Taq and HotStarTaq DNA Polymerase PCR buffer superior?
How comparable is CoralLoad gel loading dye contained in various QIAGEN PCR Kits to Sigma Red?
Have you tested the effect of inhibitors on PCR performance?
What is the fidelity of TopTaq DNA Polymerase?
What is the composition of the QIAGEN 10x PCR Buffer in Taq- and HotStarTaq DNA Polymerase Kits?
How much DNA is obtained in the average PCR reaction?
Can QIAGEN's Taq- and HotstarTaq DNA Polymerases be used for cycle sequencing?
What is the largest PCR amplicon that can be amplified with the HotStar HiFidelity Polymerase Kit?
How can one determine the optimal annealing temperature for a specific PCR assay?