QIAseq miRNA Library Kit
Gel-free miRNA Sample to Insight solution for differential expression analysis and novel discovery using next-generation sequencing
Gel-free miRNA Sample to Insight solution for differential expression analysis and novel discovery using next-generation sequencing
✓ 24/7 automatic processing of online orders
✓ Knowledgeable and professional Product & Technical Support
✓ Fast and reliable (re)-ordering
Cat. No. / ID: 331502
✓ 24/7 automatic processing of online orders
✓ Knowledgeable and professional Product & Technical Support
✓ Fast and reliable (re)-ordering
Optimized reaction chemistry enables robust, miRNA-specific libraries while minimizing reaction biases and eliminating adapter dimers. Unique Molecular Indices (UMIs) tag each miRNA at an early stage, eliminating PCR and sequencing bias. Analyze miRNA-seq data with ease using the GeneGlobe-integrated RNA-seq Analysis Portal – an intuitive, web-based data analysis solution created for biologists and included with QIAseq Stranded RNA Library Kits.
High-throughput sequencing on Illumina NovaSeq instruments is now possible with 768 unique dual indices.
Important note: We highly recommend that data is only compared with RNA-seq libraries that use the same type of indices (single or unique dual indices) in order to ensure experimental consistency. Samples and data generated with single-end indexed libraries should only be compared with other samples and data generated with single-end indices. Samples and data generated with unique dual indices should only be compared to other samples and data generated with unique dual indices.
Want to try this solution for the first time? Request a quote for a trial kit.
Mature miRNAs are naturally occurring, 22-nucleotide, non-coding RNAs that mediate posttranscriptional gene regulation. Alterations in miRNA can be correlated with gene expression changes in development, differentiation, signal transduction, infection, aging and disease. Continually growing evidence associates circulating miRNA expression with both normal and disease biology as miRNAs expressed in virtually all biofluids, including serum, plasma, cerebrospinal fluid (CSF) and urine. Specifically with cancers, numerous studies and reviews have associated the presence of various miRNAs with cancer cell proliferation, resistance to apoptosis, invasiveness and differentiation.
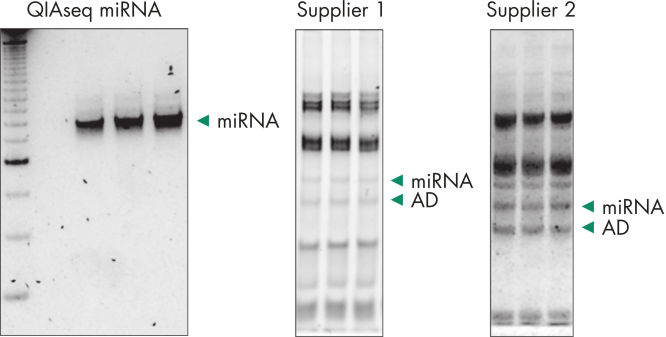
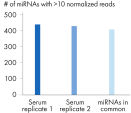
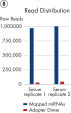
Quantification of miRNA expression can be performed using a variety of technologies including next-generation sequencing (NGS) and real-time PCR (qPCR). While NGS is the default tool for novel miRNA discovery, commercially available library preparation kits are tedious and introduce biases. As a result, qPCR has been the “go to” technology for quantification of miRNA expression, until now. QIAseq miRNA defines a new generation in small RNA sequencing products and includes several distinct features not found in other sequencing kits. With the QIAseq miRNA Library Kit, the power of NGS has been combined with single molecule quantification from UMIs to produce the most representative expression data possible.Biofluids (such as serum, plasma, CSF and urine), cells, fresh/frozen tissue and FFPE tissues containing total and miRNA are compatible with the QIAseq miRNA Library Kit. Use the QIAseq miRNA Library Kit to characterize the next circulating miRNA biomarker. Uncover miRNA signatures locked away in FFPE tissues. Completely characterize a new species. The QIAseq miRNA Library Kit has been designed to enhance yields from biofluids such as serum. In the figure Detection of miRNA, the QIAseq miRNA Library Kit shows robust detection of miRNA from serum samples. Mapped reads were then compared to adapter dimers in serum samples. QIAseq miRNA still shows superior mapping of miRNAs even with limited samples (see figure Read distribution in serum samples). With the QIAseq miRNA Library Kit, determine the differential expression of any known or novel miRNAs from any total RNA sample derived from any species.
QIAseq miRNA is the ultimate tool to enhance discovery and expression from large-scale projects with hundreds of samples down to the small pilot focused on a group of target miRNA. QIAseq miRNA offers an unrivaled Sample to Insight solution for differential expression analysis and discovery of novel miRNAs using next-generation sequencing.