

Need bulk, customized or optimized products for commercial purposes? We also offer support with logistics, compliance and more. Reach out to cooperate with QIAGEN Strategic Partnerships & OEM
Taq PCR Core Kit (1000 U)
Cat. No. / ID: 201225
4 x 250 units Taq DNA Polymerase, 10x PCR Buffer, 10x CoralLoad PCR Buffer, 5x Q-Solution, 25 mM MgCl2, dNTP Mix
Quantity
1000 U
250 U
The Taq PCR Core Kit is intended for molecular biology applications. This product is not intended for the diagnosis, prevention, or treatment of a disease.
Need bulk, customized or optimized products for commercial purposes? We also offer support with logistics, compliance and more. Reach out to cooperate with QIAGEN Strategic Partnerships & OEM
Features
- QIAGEN PCR Buffer for minimal optimization
- Additional ready-to-load PCR buffer for faster handling
- Q-Solution for amplification of GC-rich templates
- Choice of formats for convenience and ease of handling
Product Details
Taq PCR Core Kit is provided in a complete kit format comprising, Taq DNA Polymerase, the unique QIAGEN PCR Buffer that minimizes the requirement for optimization, as well as a dNTP mix. Also provided is Q-Solution, a novel additive that enables efficient amplification of "difficult" (e.g., GC rich) templates. In addition, CoralLoad PCR Buffer (containing two gel-tracking dyes) is included, enabling immediate loading of PCR products.
Performance
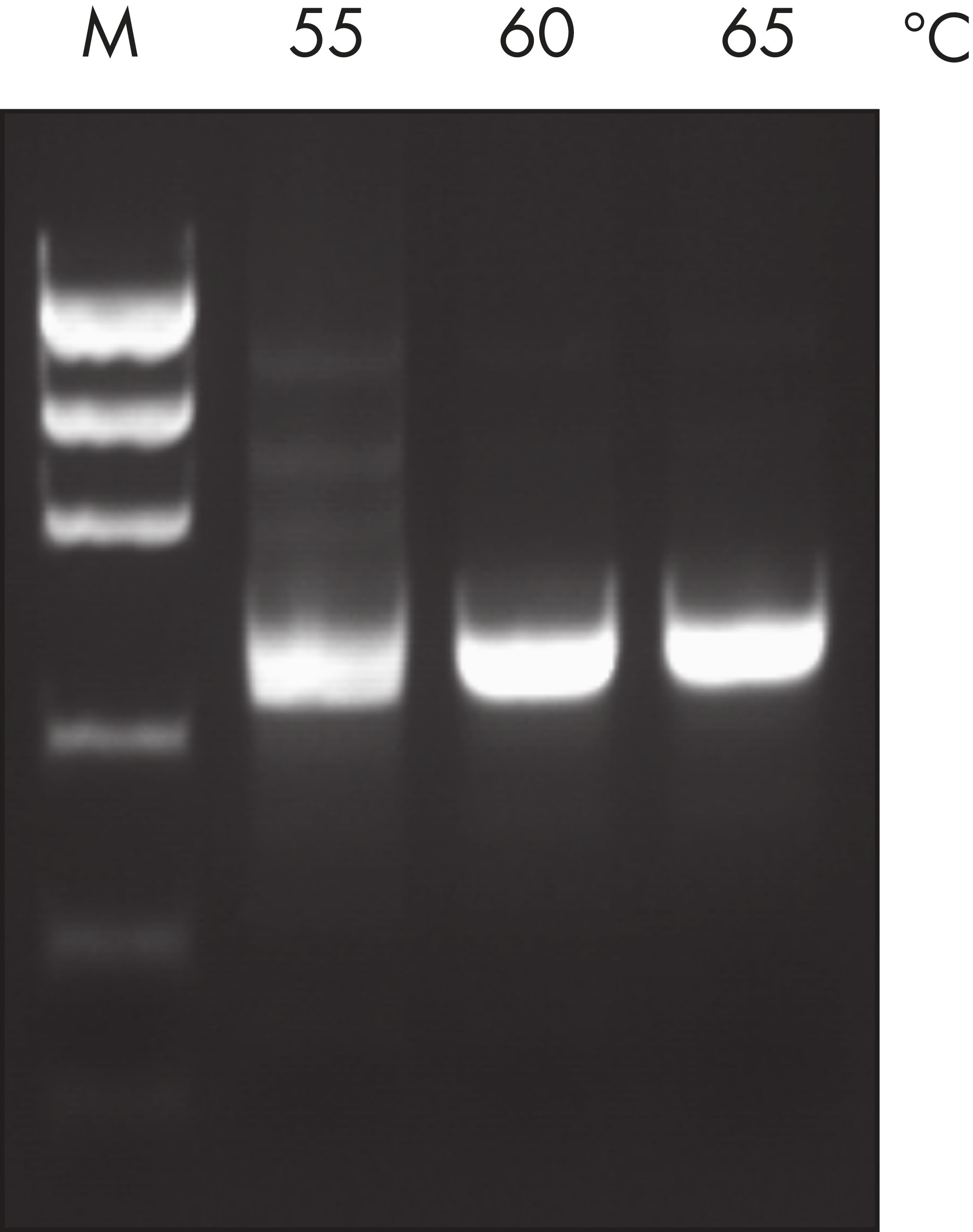
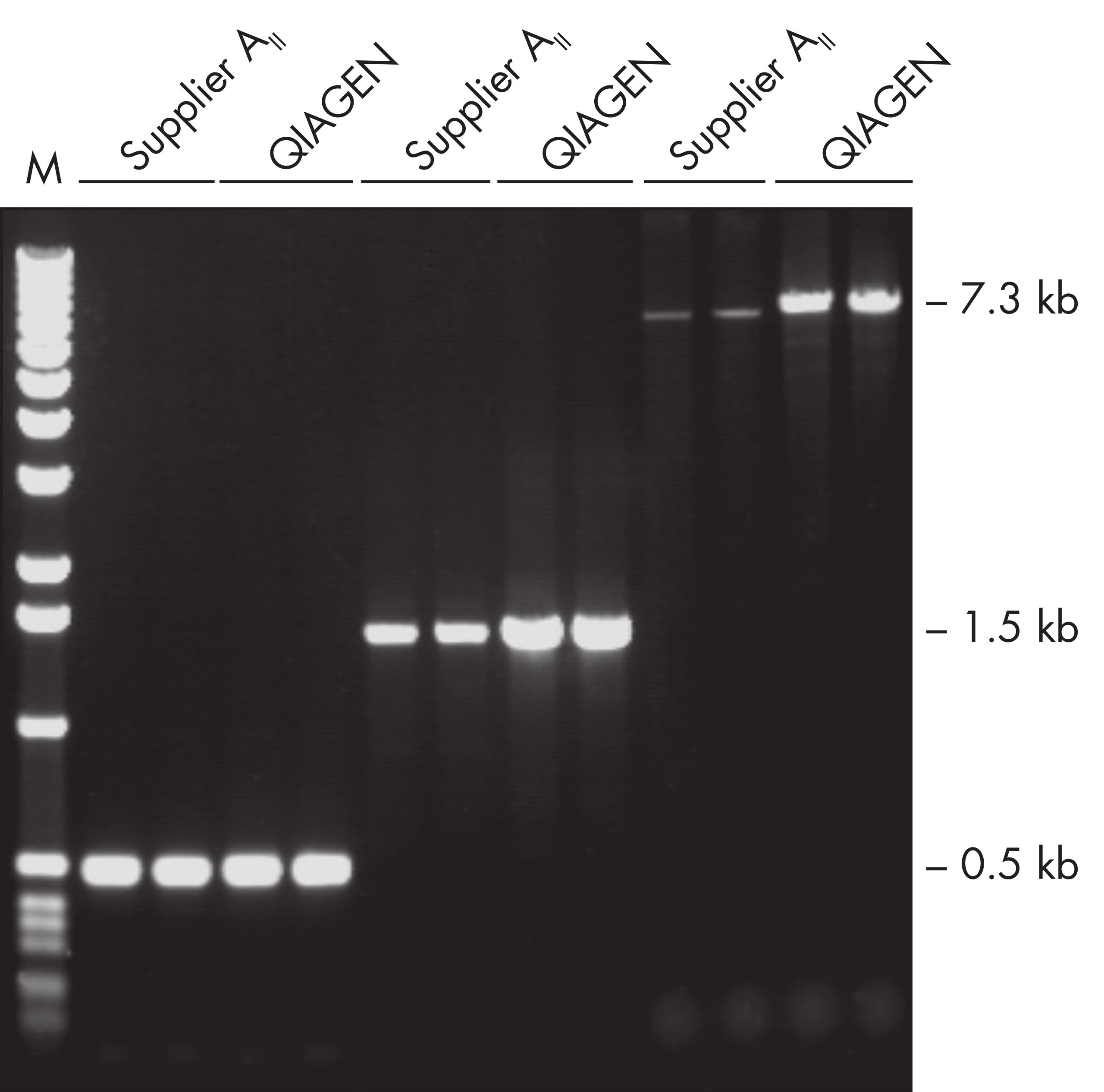
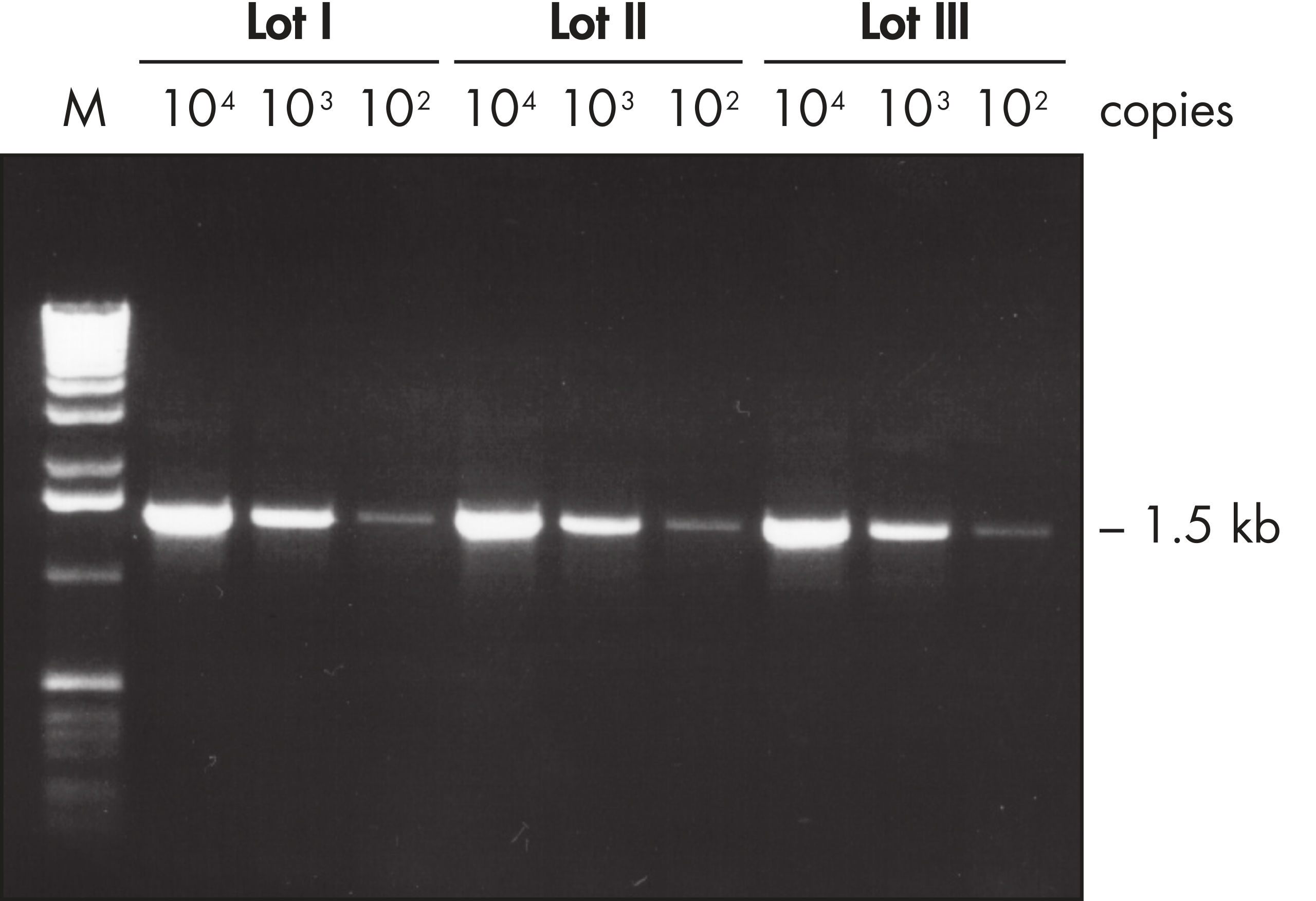
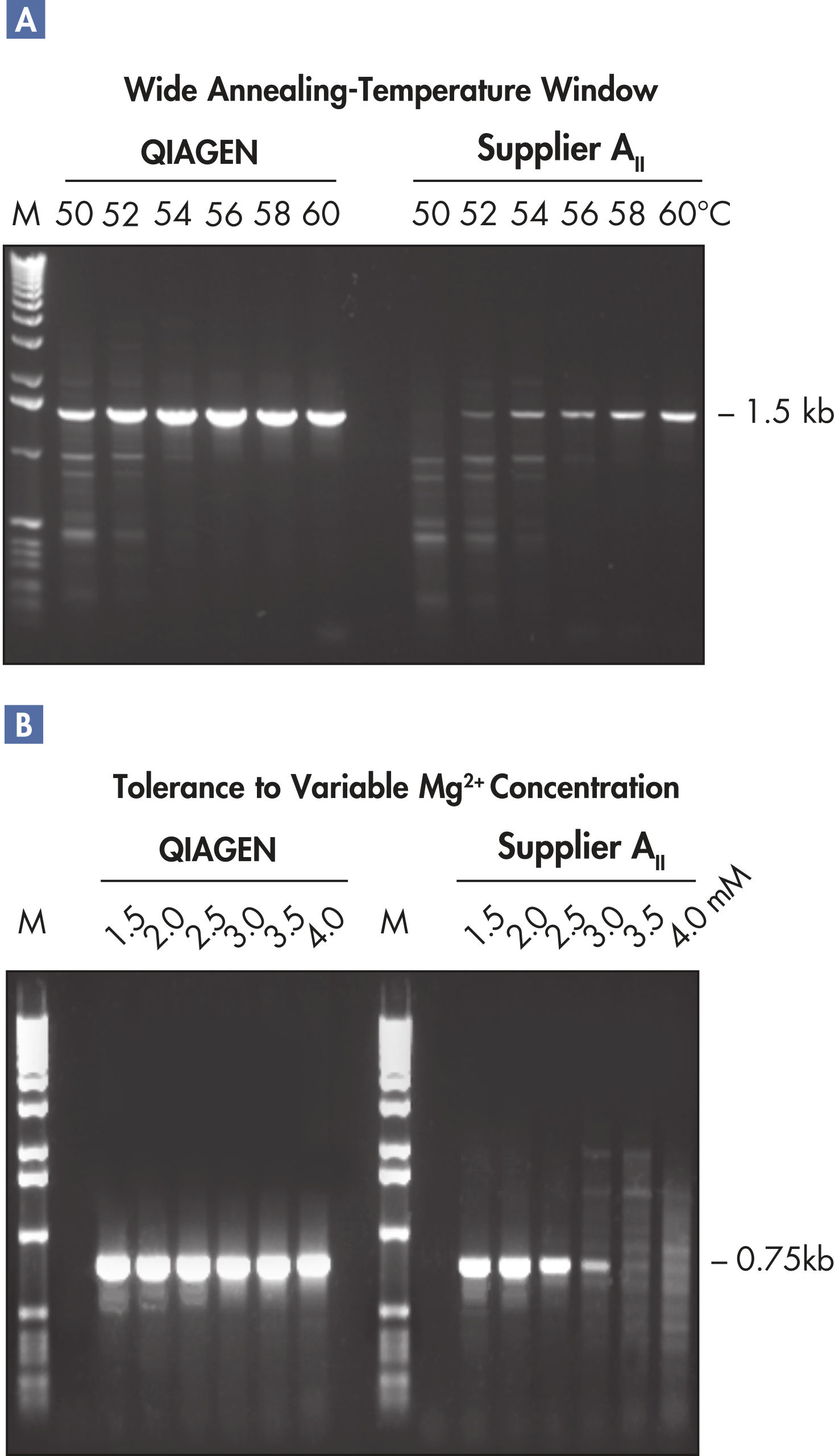
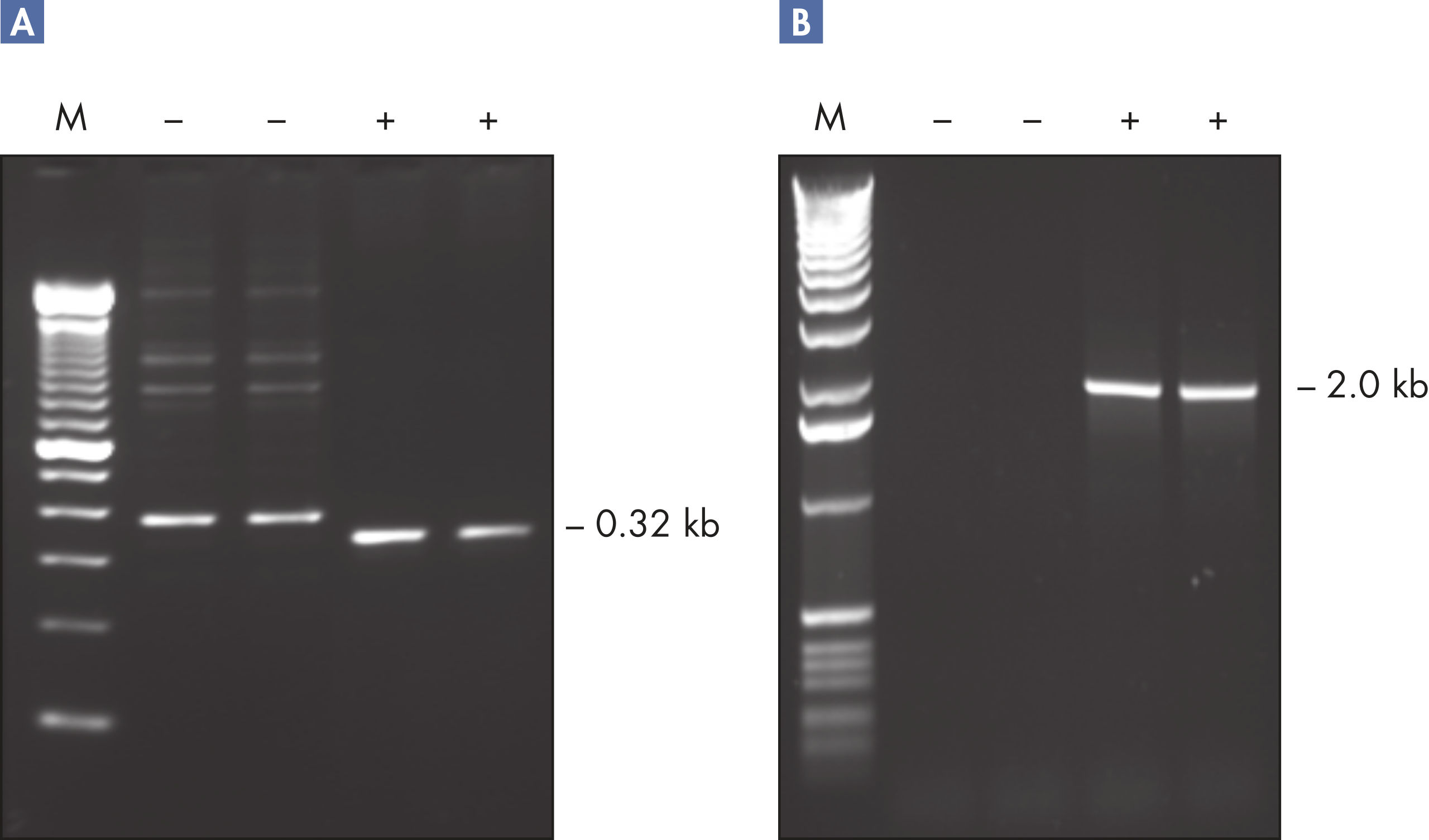
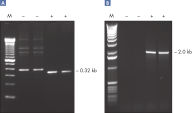
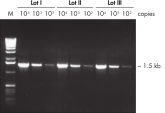
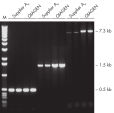
The Taq PCR Core Kit outperformed kits tested from other suppliers and delivers robust PCR performance in a wide range of PCR applications — without the need for time-consuming optimization. The kit includes Taq DNA Polymerase, a high-quality recombinant enzyme that is suitable for general and specialized PCR applications (see figures " Tolerance of different primer Tm Values" and " Specific amplification of long PCR products"). Every lot of Taq DNA Polymerase is subjected to a comprehensive range of quality control tests, including a stringent PCR specificity and reproducibility assay in which low-copy targets are amplified from human genomic DNA (see figure " Lot-to-lot reproducibility"). The unique formulation of QIAGEN PCR Buffer and CoralLoad PCR Buffer, also provided with the kit, enable highly specific PCR in a variety of PCR conditions with minimal optimization requirements (see figures " Wide annealing-temperature window" and " Tolerance to variable magnesium concentration"). In addition, CoralLoad PCR Buffer enables immediate loading of PCR products onto an agarose gel for even easier handling and faster results. Suboptimal PCR can be improved using Q-Solution, a PCR additive, also provided with the kit (see figure " Amplification of difficult templates").
Taq DNA Polymerase specifications
Concentration: 5 units/µl
Recombinant enzyme: Yes
Substrate analogs: dNTP, ddNTP, dUTP, biotin-11-dUTP, DIG-11-dUTP, fluorescent-dNTP/ddNTP
Extension rate: 2–4 kb/min at 72°C
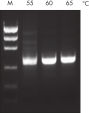
Half-life: 10 min at 97°C; 60 min at 94°C
Amplification efficiency: ≥105 fold
5'–>3' exonuclease activity: Yes
Extra A addition: Yes
3'–>5' exonuclease activity: No
Contaminating nucleases: No
Contaminating RNases: No
Contaminating proteases: No
Self-priming activity: No >
See figures
Principle
The Taq PCR Core Kit includes everything required for convenient and reliable PCR —Taq DNA Polymerase, QIAGEN PCR Buffer, CoralLoad PCR Buffer, Q-Solution, dNTP Mix, and MgCl2.
Taq DNA Polymerase
Taq DNA Polymerase is a high-quality recombinant enzyme that is suitable for general and specialized PCR applications (see figures " Tolerance of different primer Tm values" and " Specific amplification of long PCR products").
QIAGEN PCR Buffer
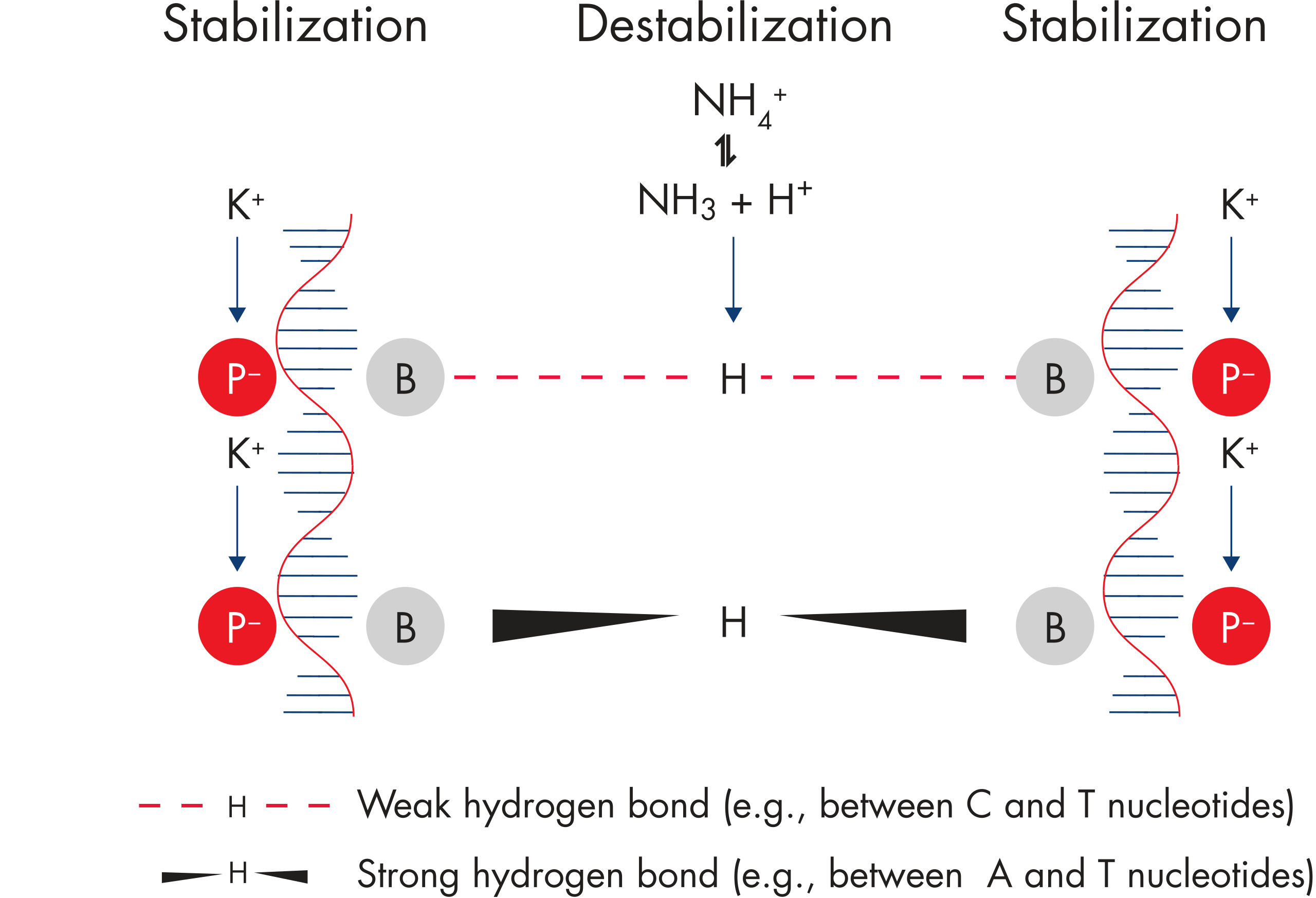
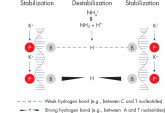
The innovative QIAGEN PCR Buffer has been developed to save time and effort by reducing the need for PCR optimization. QIAGEN PCR Buffer contains both KCl and (NH4)2SO4 (see figure " Increased specificity of primer annealing"). This unique buffer facilitates the amplification of specific PCR products. During the annealing step of every PCR cycle, the buffer allows a high ratio of specific-to-nonspecific primer binding. Owing to a uniquely balanced combination of KCl and (NH4)2SO4, the PCR buffer provides stringent primer-annealing conditions over a wider range of annealing temperatures and Mg2+ concentrations than conventional PCR buffers. Optimization of PCR by varying the annealing temperature or the Mg2+ concentration is dramatically reduced and often not required (see figures " Wide annealing temperature window" and " Tolerance to variable magnesium concentration").
CoralLoad PCR Buffer
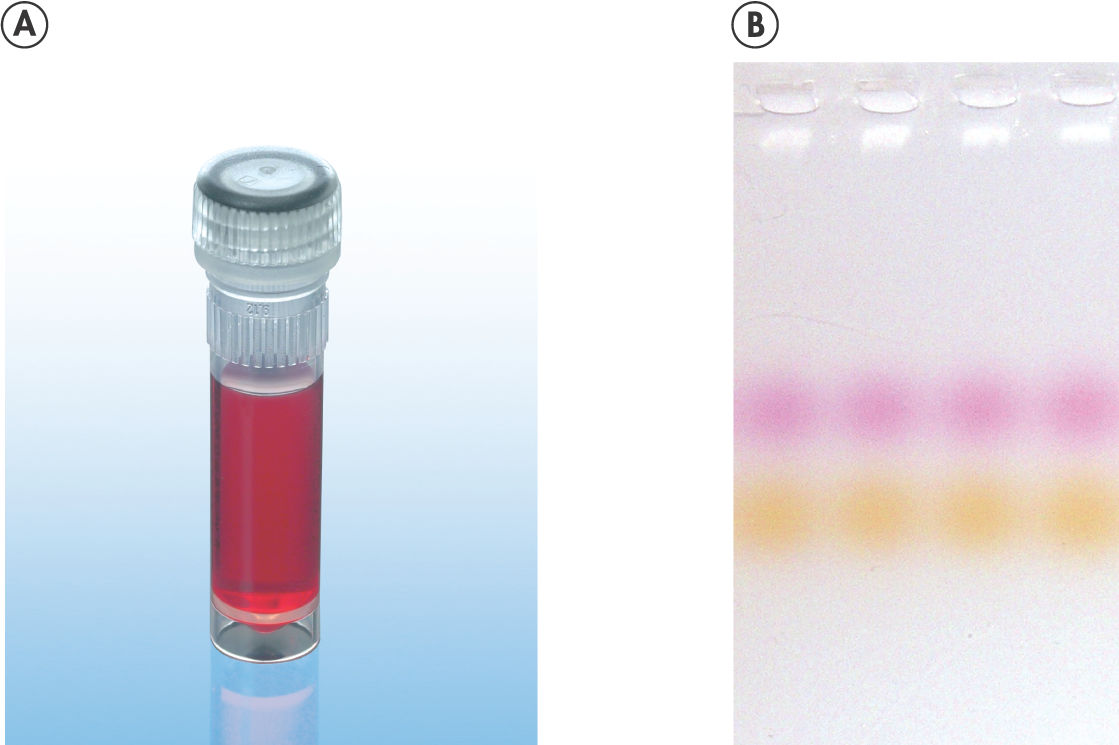
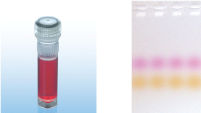
CoralLoad PCR Buffer has all the advantages of QIAGEN PCR Buffer. In addition, it can also be used to directly load the PCR reaction onto an agarose gel — separate addition of a gel loading buffer is not required. CoralLoad PCR Buffer provides the same high PCR specificity and minimal reaction optimization as the conventional QIAGEN PCR Buffer. Additionally, it contains two marker dyes — an orange dye and a red dye — that facilitate estimation of DNA migration distance and optimization of agarose gel run time (see figure " CoralLoad PCR Buffer"). The buffer ensures improved pipetting visibility and enables direct loading of PCR products onto a gel, for enhanced convenience.
Q-Solution
Q-Solution facilitates amplification of GC-rich templates or templates with a high degree of secondary structure by modifying the melting behavior of DNA. Use of this unique reagent often enables or improves suboptimal PCR (see figure " Amplification of difficult templates"). Unlike DMSO and other PCR additives, Q-Solution is used at a defined working concentration with any primer–template system and is not toxic.
See figures
Procedure
The Taq PCR Core Kit provides all the components required to set up a PCR and can be used for a multitude of PCR-based applications. The optimized, easy-to-follow, streamlined protocol provided with the kit ensures successful PCR results. Suboptimal PCR is simplified with Q-Solution, a unique PCR additive, also included with the kit.
Applications
Taq DNA Polymerase is used for standard and specialized applications, including:
- General PCR
- RT-PCR
- Screening
- PCR-based DNA fingerprinting (VNTR, STR, and RAPD)
Supporting data and figures
CoralLoad PCR Buffer.
CoralLoad PCR Buffer.

Specifications
| Features | Specifications |
|---|---|
| Applications | PCR, RT-PCR, DNA fingerprinting |
| Sample/target type | Genomic DNA and cDNA |
| Real-time or endpoint | Endpoint |
| dNTP's included | Yes |
| Single or multiplex | Single |
| Reaction type | PCR amplification |
| Mastermix | No |
| With/without hotstart | Without hotstart |
| Enzyme activity | 5' -> 3' exonuclease activity |
Resources
Brochures & Guides (4)
Quick-Start Protocols (1)
Safety Data Sheets (2)
Kit Handbooks (1)
Technical Information (1)
Certificates of Analysis (1)
Publications
Nonhematopoietic/endothelial SSEA-1+ cells define the most primitive progenitors in the adult murine bone marrow mesenchymal compartment.
Blood; 2006; 109 (3):1298-306 2006 Sep 26 PMID:17003364
Combined real-time PCR and rpoB gene pyrosequencing for rapid identification of Mycobacterium tuberculosis and determination of rifampin resistance directly in clinical specimens.
J Clin Microbiol; 2010; 48 (4):1182-8 2010 Jan 27 PMID:20107097
Expression of calcium channels along the differentiation of cultured trophoblast cells from human term placenta.
Biol Reprod; 2002; 67 (5):1473-9 2002 Nov PMID:12390878
An immune evasion mechanism for spirochetal persistence in Lyme borreliosis.
J Exp Med; 2002; 195 (4):415-22 2002 Feb 18 PMID:11854355
FAQ
What should the starting template DNA quality and quantity be for PCR?
What kind of PCR products can be cloned with the QIAGEN PCR Cloning Kit?
Does QIAGEN sell Q-Solution separately?
Do you have a protocol for polyacrylamide gel analysis of oligonucleotides?
Can Taq DNA Polymerase use RNA as a template, and generate false positives in "no-RT" controls?
Is Q-Solution required for PCR with QIAGEN's PCR kits?
How comparable is CoralLoad gel loading dye contained in various QIAGEN PCR Kits to Sigma Red?
Have you tested the effect of inhibitors on PCR performance?
What is the composition of the QIAGEN 10x PCR Buffer in Taq- and HotStarTaq DNA Polymerase Kits?
How much DNA is obtained in the average PCR reaction?
Can QIAGEN's Taq- and HotstarTaq DNA Polymerases be used for cycle sequencing?
How can one determine the optimal annealing temperature for a specific PCR assay?