

✓ 24/7 automatic processing of online orders
✓ Knowledgeable and professional Product & Technical Support
✓ Fast and reliable (re)-ordering
QIAseq Methyl Library Kit (24)
Cat. No. / ID: 180502
Enzymes, buffers, adapters (up to 24 sample indexes) and QIAseq Beads for construction of 24 libraries from bisulfite-treated DNA
Log in To see your account pricing.
✓ 24/7 automatic processing of online orders
✓ Knowledgeable and professional Product & Technical Support
✓ Fast and reliable (re)-ordering
Features
- High library yield and mapping rates
- Low bias and error rates
- Wide range of DNA input
- Compatible with cfDNA samples
- Suitable for enriched fragmented samples (e.g., RRBS, MeDIP)
Product Details
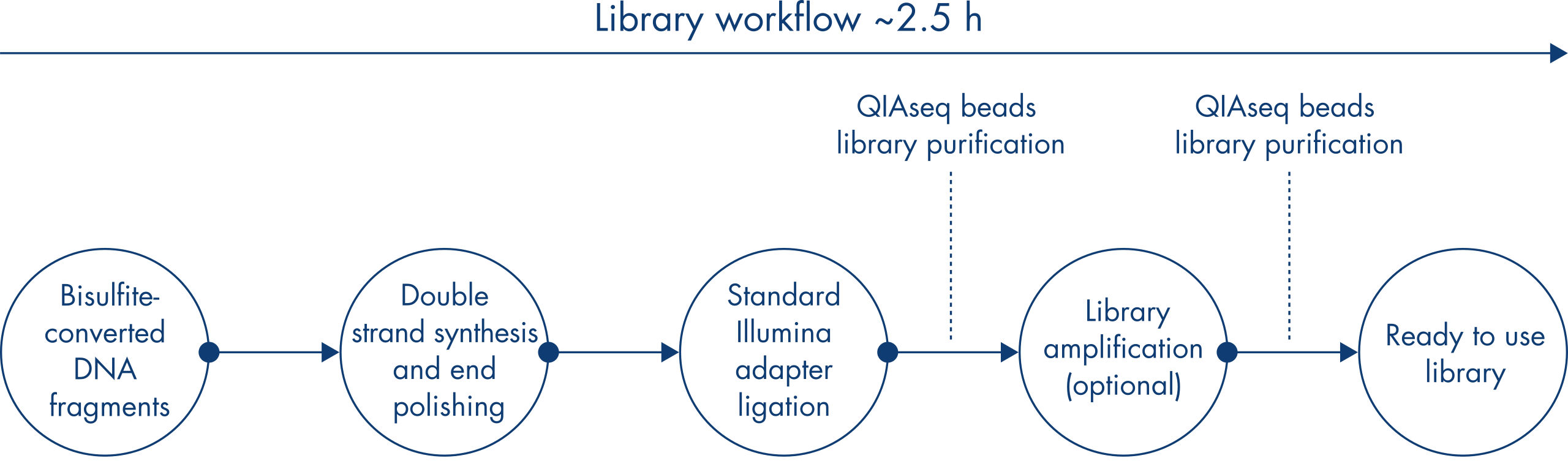
The QIAseq Methyl Library Kit is a library construction kit used for preparing libraries from bisulfite-treated DNA samples for whole genome methylation studies using Illumina platforms. It is developed for researchers who need a robust solution to interrogate methylation events in their biological samples. The kit combines several library preparation reactions into one streamlined step that enables complete library preparation in 2.5 hours and allows the full workflow, including bisulfite conversion, to be performed in less than a day. Unlike other kits that require considerable DNA input, the QIAseq Methyl Library kit allows DNA inputs as low as 100 pg to deliver high-quality, high-yield libraries for use with Illumina platforms.
The kit includes all required components to go from bisulfite-treated DNA to libraries: enzymes, buffers and reagents for double-strand synthesis, end-polishing of bisulfite-converted DNA, ligation of Illumina adapters, library purification with QIAseq Beads and library amplification with the VeraSeq ULtra DNA Polymerase.
Performance
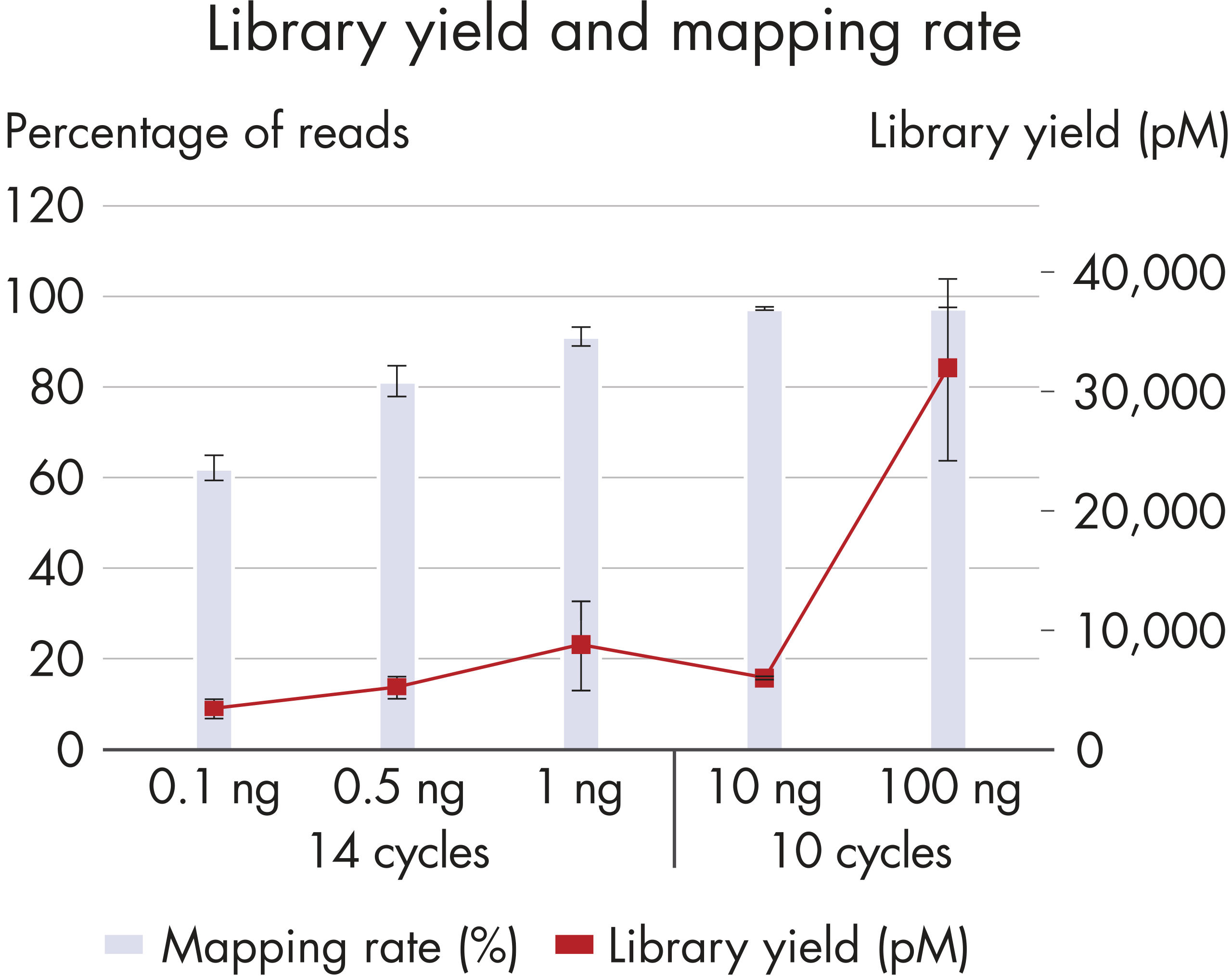
High yield
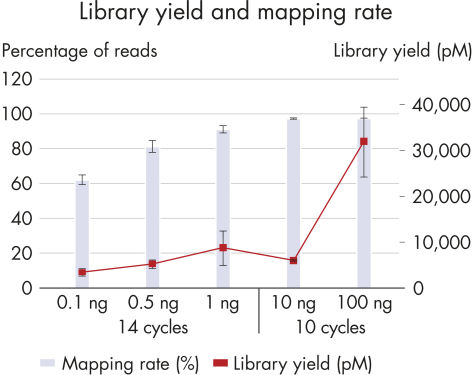
The superior chemistry of the QIAseq Methyl Library Kit enables high library yields, using as little as 100 pg of input DNA for library construction, from a low number of amplification cycles (Figure High library yield and mapping rate).High mapping rates
One of the major challenges in the use of NGS for methylation analysis is low mapping rates, resulting in inefficient use of the sequencing run. The QIAseq Methyl Library Kit overcomes this challenge by delivering mapping rates greater than 90%, with an optimal DNA input range of 1–100 ng (Figure High library yield and mapping rate).Interrogates CpG methylation with low bias
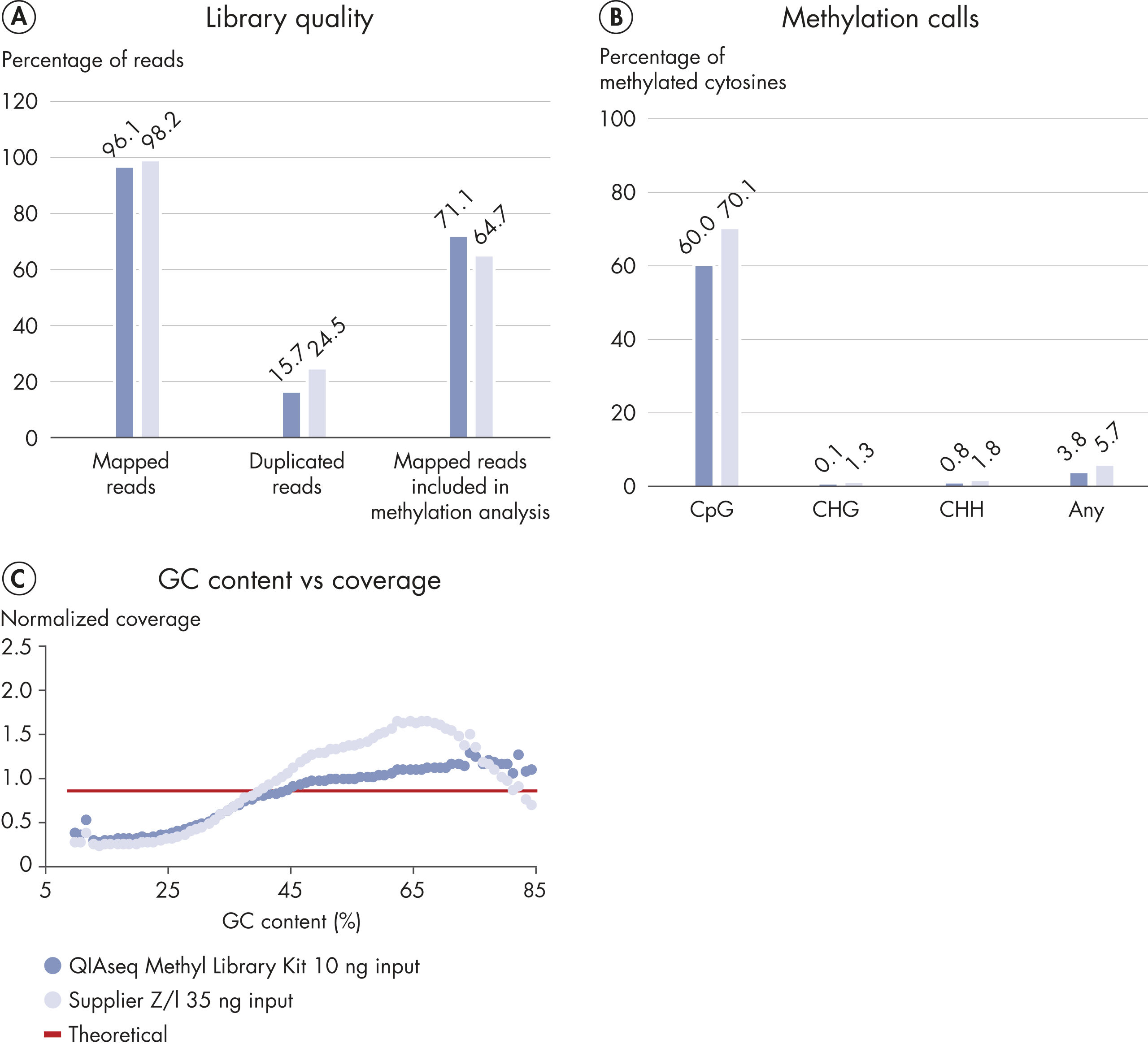
Accurate methylation analysis is mostly affected by the input material and unbiased conversion of the bisulfite-treated and heavily-fragmented DNA into library molecules. The high degree of bisulfite conversion provided by EpiTect Fast and efficient repair of the bisulfite-treated DNA, coupled with efficient ligation of adapters, make accurate methylation analysis possible, even from very low DNA input (Figure Accurate methylation state calling).Low error rates
With an optimal input range of 1–100 ng DNA, the QIAseq Methyl Library Kit, which includes the high-fidelity VeraSeq ULtra DNA Polymerase, reduces error rates for more accurate assessment of methylation events.See figures
Principle
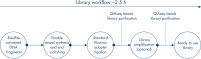
The QIAseq Methyl Library Kit contains enzyme mixes and buffers for preparing bisulfite-treated single-stranded DNA for ligation of adapters that allow sequencing on Illumina platforms and amplification of the libraries. Magnetic beads are included for library purification. In a simplified workflow, the kit uses an enzyme mix and random short primers for double strand synthesis of single-stranded bisulfite-treated DNA. In the same reaction setup, enzymes will end-polish and prepare the ends of the fragments for subsequent adapter ligation. The reactions, double-strand synthesis, DNA end-polishing and adapter ligation, take place in the same tube, thereby minimizing sample loss. The ligation step is followed by a purification step using the included QIAseq Beads. Library amplification is performed using the high-fidelity VeraSeq ULtra DNA Polymerase, which is an engineered, ultra-thermostable, uracil-literate polymerase designed to maximize the speed, accuracy and length of DNA synthesis during sequencing template preparation. The result is a novel enzyme that can read through uracil, extend a kilobase of sequence in 15 seconds and has an accuracy 25 times higher than Taq DNA Polymerase. This enables the amplification of WGBS libraries without sequence bias.
Dual-bar-coded, plate-format adapters are included in QIAseq Methyl DNA Library Kits. Each adapter well contains a single-use adapter consisting of a unique combination of two eight-nucleotide identification bar codes. By combining one of eight D5 bar codes and one of three D7 bar codes in each ready-to-use adapter, this kit supports up to 24-plexing prior to sequencing (Figure The QIAseq Methyl Library Kit workflow).
Dual-bar-coded, plate-format adapters are included in QIAseq Methyl DNA Library Kits. Each adapter well contains a single-use adapter consisting of a unique combination of two eight-nucleotide identification bar codes. By combining one of eight D5 bar codes and one of three D7 bar codes in each ready-to-use adapter, this kit supports up to 24-plexing prior to sequencing (Figure The QIAseq Methyl Library Kit workflow).
See figures
Procedure
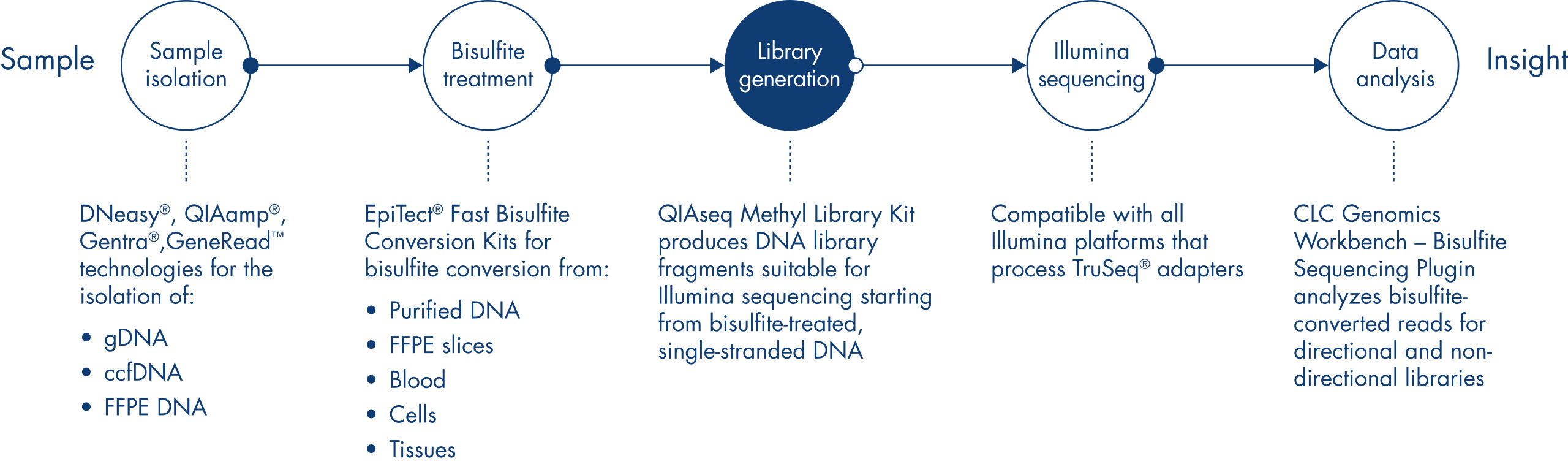
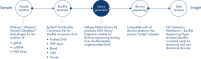
Before library construction, DNA samples undergo bisulfite treatment with dedicated EpiTect Fast protocols for the complete conversion of non-methylated cytosines. Library construction begins with double-strand synthesis and end-polishing followed by adapter ligation. Following library construction, the reaction cleanup and removal of adapter-dimers is achieved using QIASeq Beads. After library amplification, a final cleanup step is performed with QIASeq Beads. After sequencing on an Illumina platform, bioinformatic analysis is simplified by using the Bisulfite Sequencing plugin (version 2.0 or later) for Biomedical Genomics Workbench and CLC Genomics Workbench for non-directional libraries (Figure Sample to Insight workflow for the analysis of methylation events).
See figures
Applications
The QIAseq Methyl Library Kit produces high-quality libraries for the analysis of CpG methylation events from a wide range of samples, including gDNA, cfDNA and FFPE, as well as fragmented and enriched CpG fragments.
Bisulfite conversion leads to DNA damage and makes bisulfite sequencing of heavily fragmented samples very difficult.
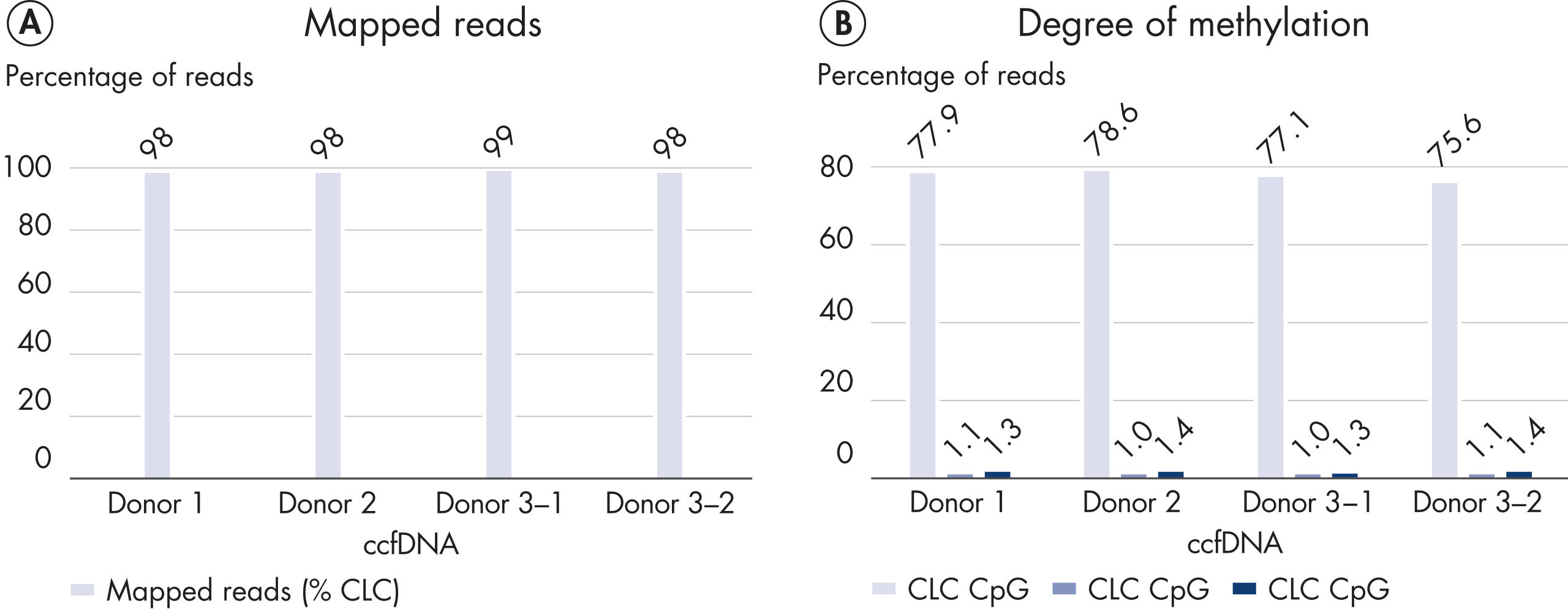
The robust chemistry of the EpiTect Fast Kits and optimized protocols of the QIAseq Methyl Library Kit allow sequencing of difficult samples such as ccfDNA (Figure High percentage of mapped reads and accurate methylation calls with low-input circulating cell-free DNA).
Bisulfite conversion leads to DNA damage and makes bisulfite sequencing of heavily fragmented samples very difficult.
The robust chemistry of the EpiTect Fast Kits and optimized protocols of the QIAseq Methyl Library Kit allow sequencing of difficult samples such as ccfDNA (Figure High percentage of mapped reads and accurate methylation calls with low-input circulating cell-free DNA).
See figures
Supporting data and figures
High library yield and mapping rate.
A wide range of DNA inputs from Jurkat cells were bisulfite-treated using a dedicated EpiTect Fast protocol and libraries were prepared using the QIAseq Methyl Library Kit and sequenced on a MiSeq instrument. Enough library to load onto a MiSeq was generated from as little as 100 pg with high mapping rates.



Resources
Safety Data Sheets (1)
Quick-Start Protocols (1)
Kit Handbooks (1)
Certificates of Analysis (1)